

DART-ID increases single-cell proteome coverage
- PMID: 31260443
- PMCID: PMC6625733
- DOI: 10.1371/journal.pcbi.1007082
DART-ID increases single-cell proteome coverage
Abstract
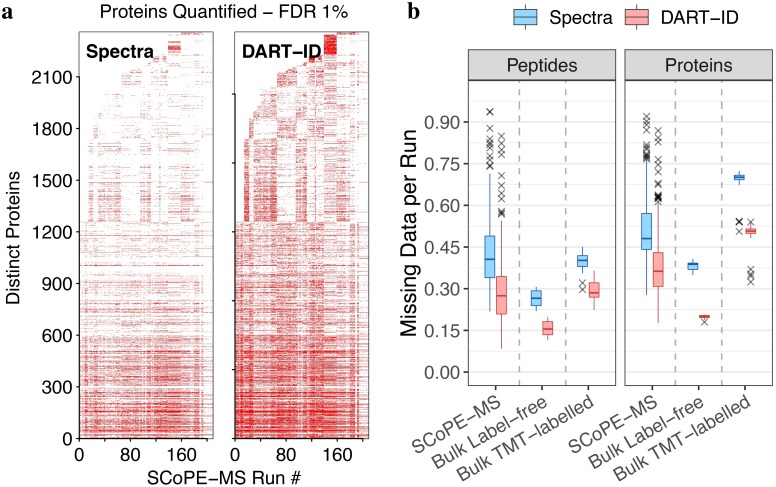
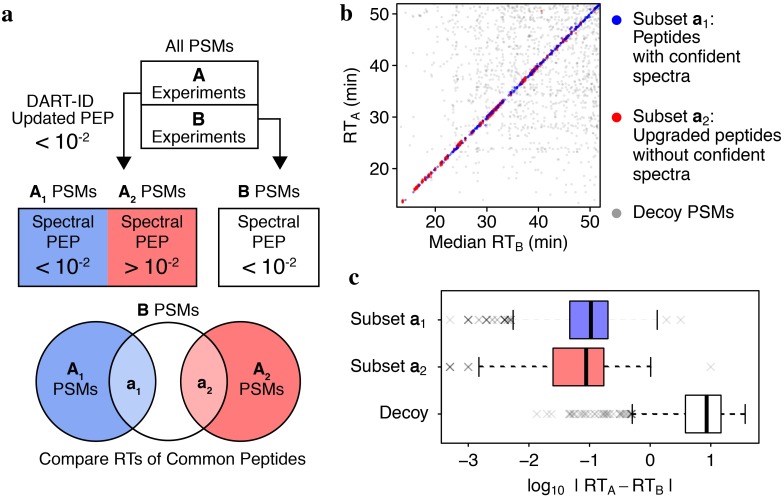
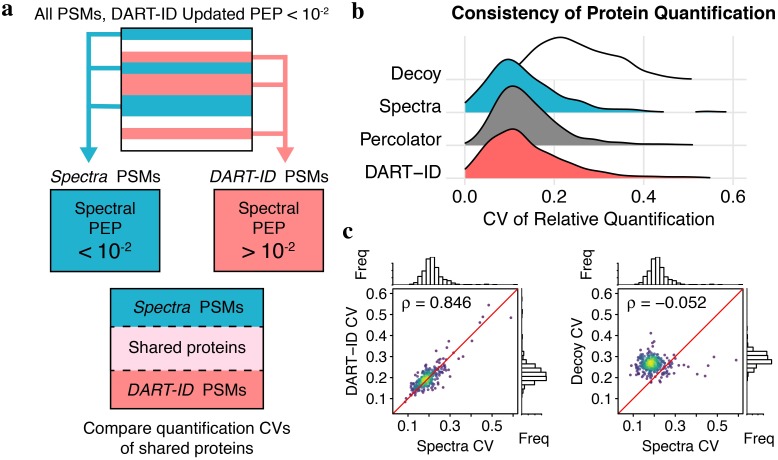
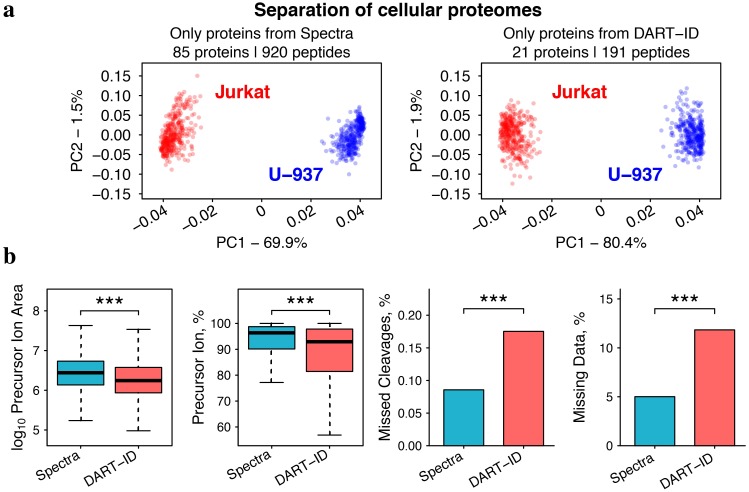
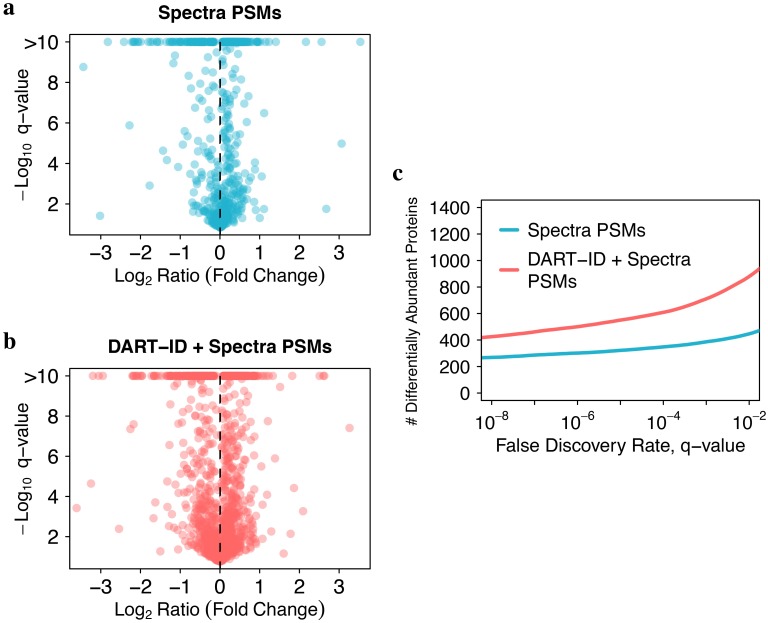
Analysis by liquid chromatography and tandem mass spectrometry (LC-MS/MS) can identify and quantify thousands of proteins in microgram-level samples, such as those comprised of thousands of cells. This process, however, remains challenging for smaller samples, such as the proteomes of single mammalian cells, because reduced protein levels reduce the number of confidently sequenced peptides. To alleviate this reduction, we developed Data-driven Alignment of Retention Times for IDentification (DART-ID). DART-ID implements principled Bayesian frameworks for global retention time (RT) alignment and for incorporating RT estimates towards improved confidence estimates of peptide-spectrum-matches. When applied to bulk or to single-cell samples, DART-ID increased the number of data points by 30-50% at 1% FDR, and thus decreased missing data. Benchmarks indicate excellent quantification of peptides upgraded by DART-ID and support their utility for quantitative analysis, such as identifying cell types and cell-type specific proteins. The additional datapoints provided by DART-ID boost the statistical power and double the number of proteins identified as differentially abundant in monocytes and T-cells. DART-ID can be applied to diverse experimental designs and is freely available at http://dart-id.slavovlab.net.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
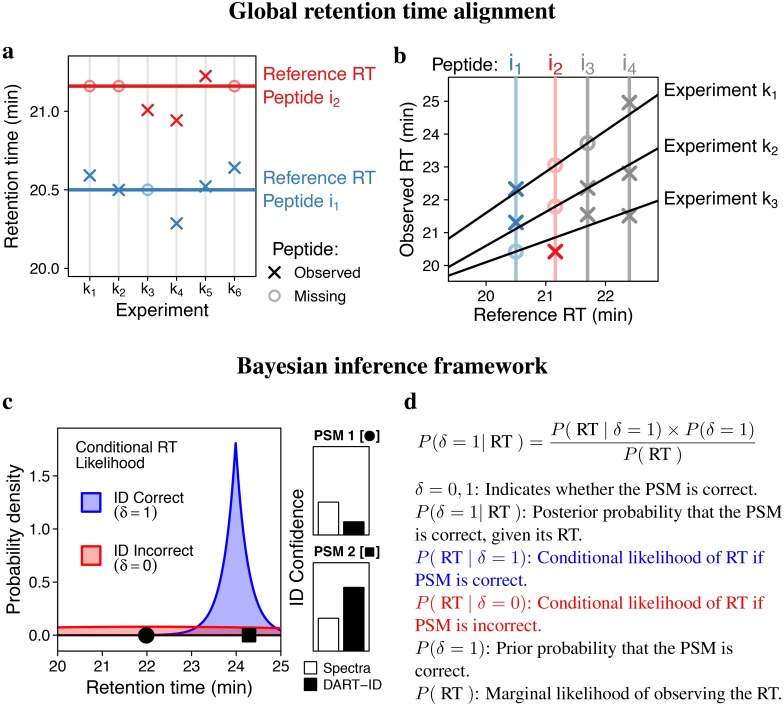
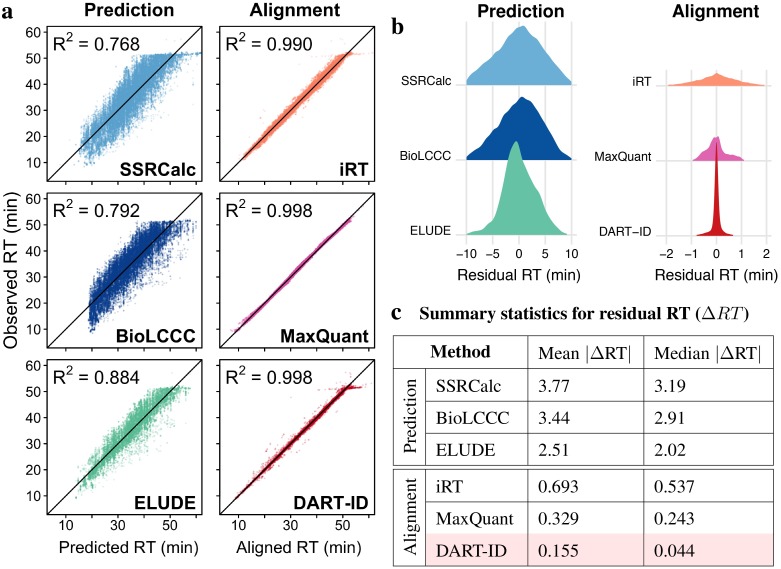
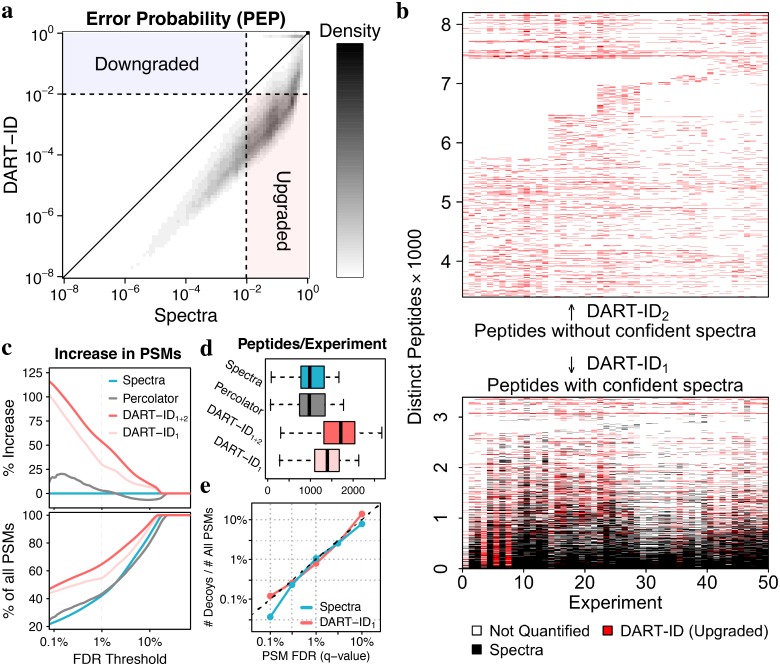
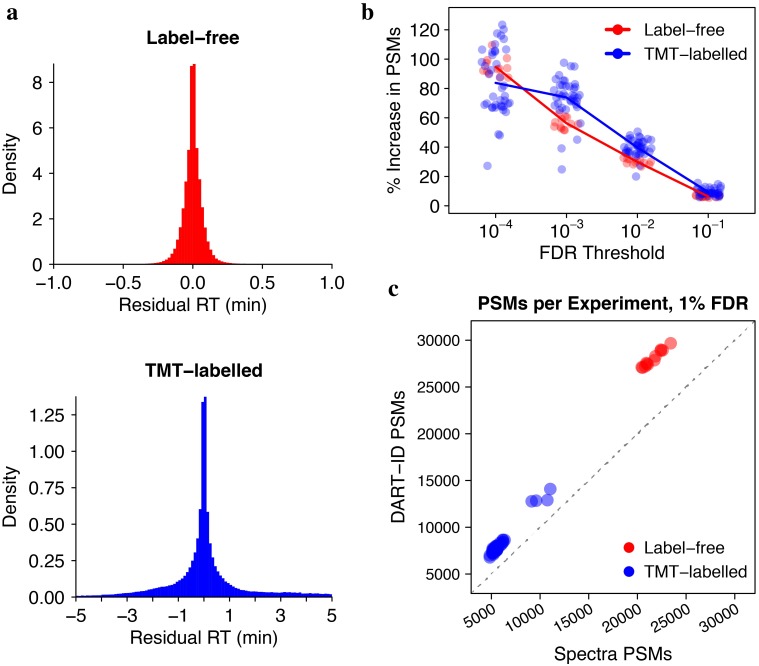
References
-
- Specht H, Harmange G, Perlman DH, Emmott E, Niziolek Z, Budnik B, et al. Automated sample preparation for high-throughput single-cell proteomics. bioRxiv. 2018. 10.1101/399774 - DOI
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
