

Rewiring of the Transcription Factor Network in Acute Myeloid Leukemia
- PMID: 31263370
- PMCID: PMC6595639
- DOI: 10.1177/1176935119859863
Rewiring of the Transcription Factor Network in Acute Myeloid Leukemia
Abstract
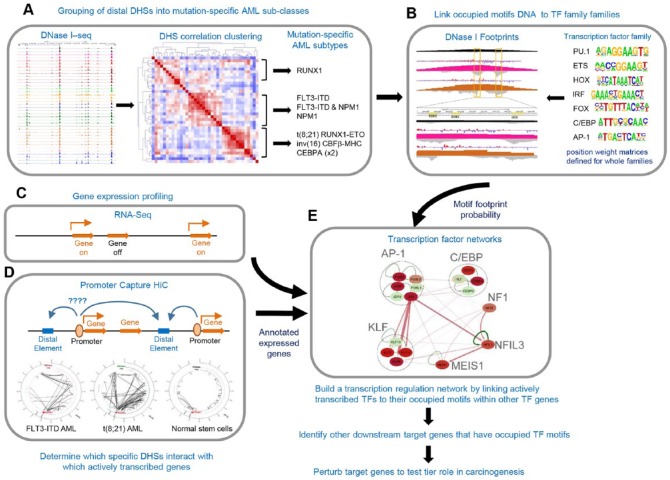
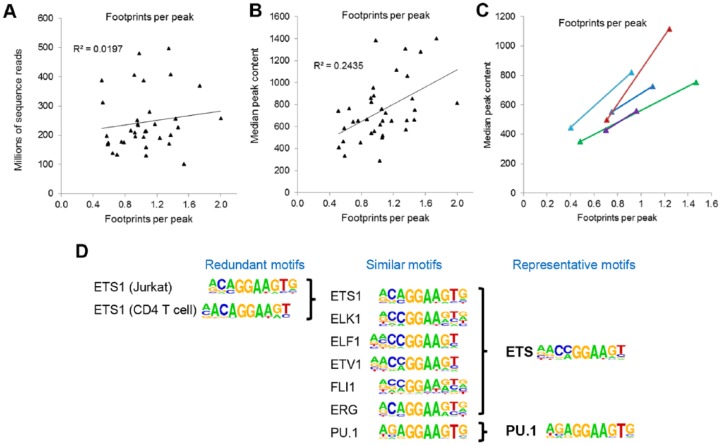
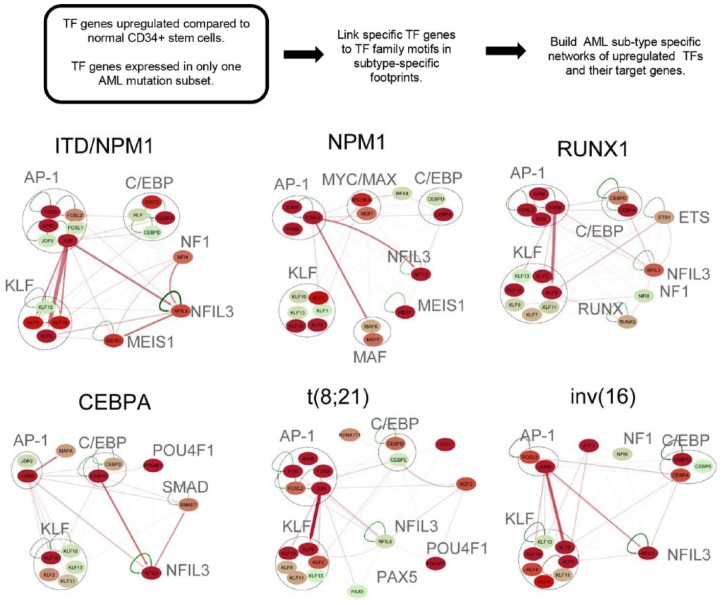
Acute myeloid leukemia (AML) is a highly heterogeneous cancer associated with different patterns of gene expression determined by the nature of their DNA mutations. These mutations mostly act to deregulate gene expression by various mechanisms at the level of the nucleus. By performing genome-wide epigenetic profiling of cis-regulatory elements, we found that AML encompasses different mutation-specific subclasses associated with the rewiring of the gene regulatory networks that drive differentiation into different directions away from normal myeloid development. By integrating epigenetic profiles with gene expression and chromatin conformation data, we defined pathways within gene regulation networks that were differentially rewired within each mutation-specific subclass of AML. This analysis revealed 2 major classes of AML: one class defined by mutations in signaling molecules that activate AP-1 via the mitogen-activated protein (MAP) kinase pathway and a second class defined by mutations within genes encoding transcription factors such as RUNX1/CBFβ and C/EBPα. By identifying specific DNA motifs protected from DNase I digestion at cis-regulatory elements, we were able to infer candidate transcription factors bound to these motifs. These integrated analyses allowed the identification of AML subtype-specific core regulatory networks that are required for AML development and maintenance, which could now be targeted in personalized therapies.
Keywords: AP-1; Acute myeloid leukemia; CEBPA; DNA mutation; DNase; FLT3; RUNX1; gene expression; gene regulation; network; transcription factor.
Conflict of interest statement
Declaration of Conflicting Interests:The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Figures
Comment on
-
Subtype-specific regulatory network rewiring in acute myeloid leukemia.Nat Genet. 2019 Jan;51(1):151-162. doi: 10.1038/s41588-018-0270-1. Epub 2018 Nov 12. Nat Genet. 2019. PMID: 30420649 Free PMC article.
References
-
- Cockerill PN. Structure and function of active chromatin and DNase I hypersensitive sites. FEBS J. 2011;278:2182–2210. - PubMed
-
- Bonifer C, Cockerill PN. Chromatin structure profiling identifies crucial regulators of tumor maintenance. Trends Cancer. 2015;1:157–160. - PubMed
-
- Robertson G, Hirst M, Bainbridge M, et al. Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat Methods. 2007;4:651–657. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
