

Resistance to the Tat Inhibitor Didehydro-Cortistatin A Is Mediated by Heightened Basal HIV-1 Transcription
- PMID: 31266880
- PMCID: PMC6606815
- DOI: 10.1128/mBio.01750-18
Resistance to the Tat Inhibitor Didehydro-Cortistatin A Is Mediated by Heightened Basal HIV-1 Transcription
Abstract
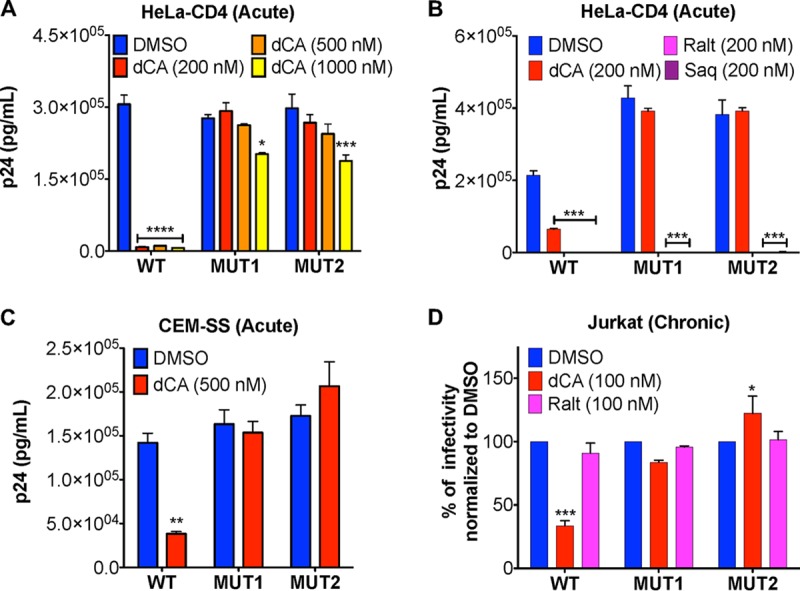
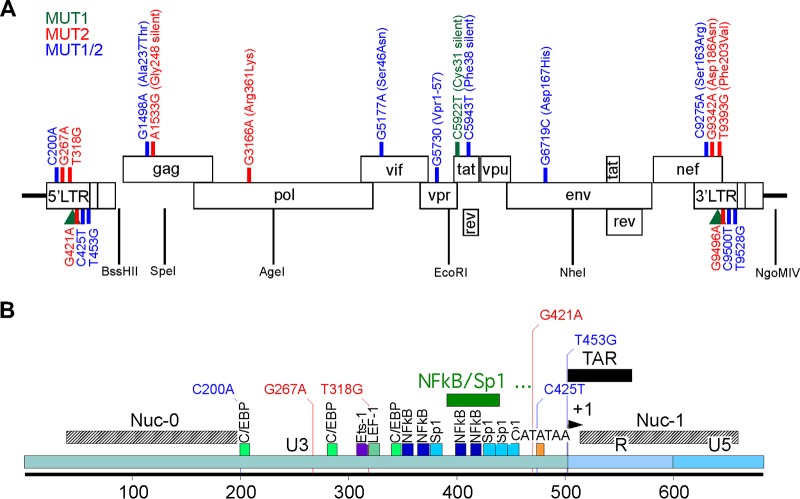
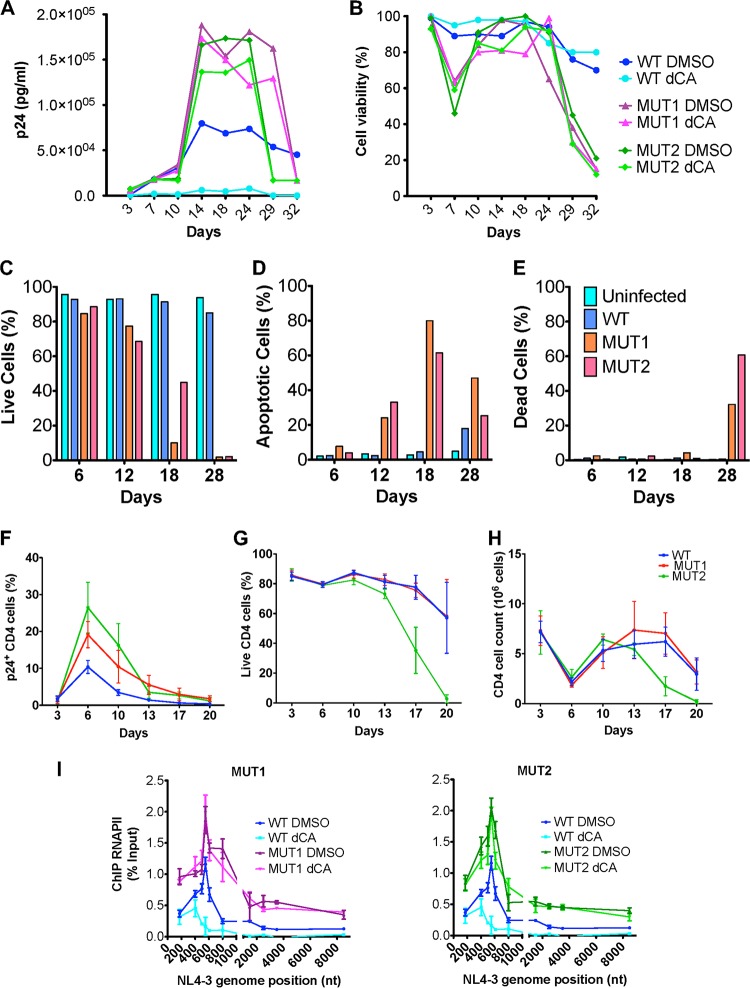
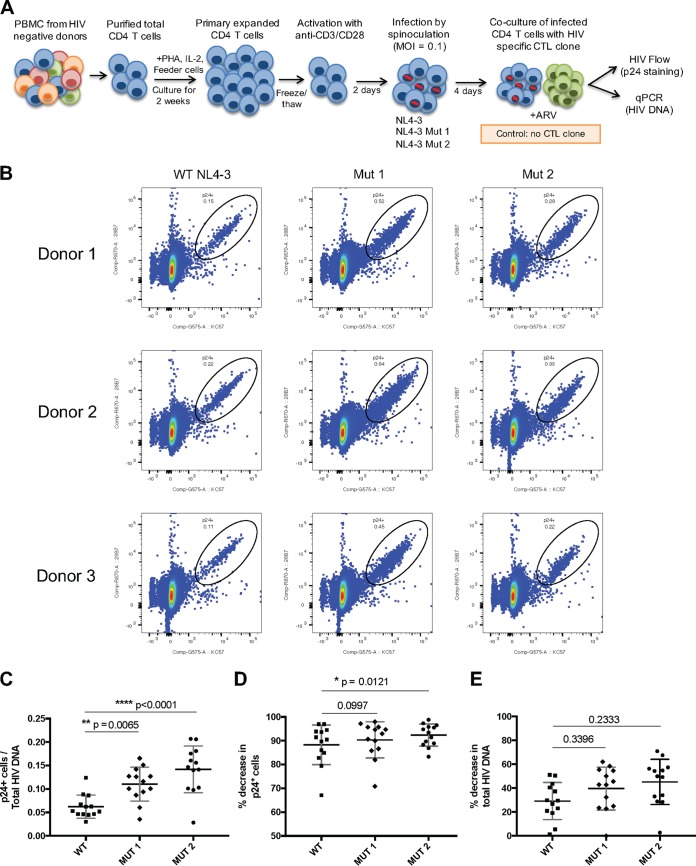
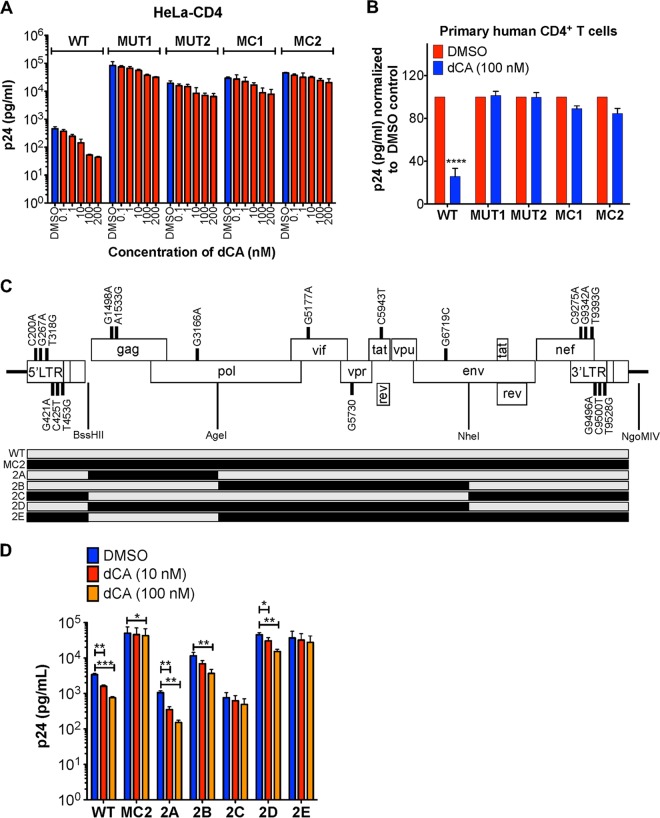
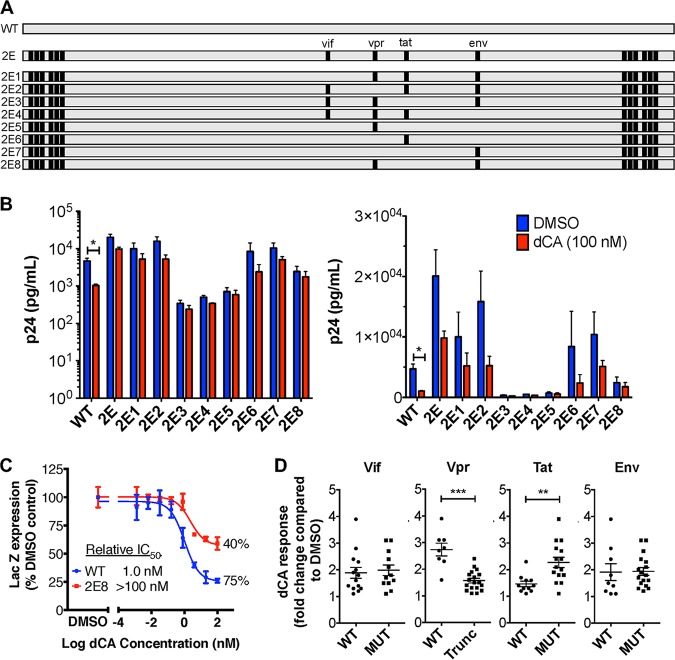
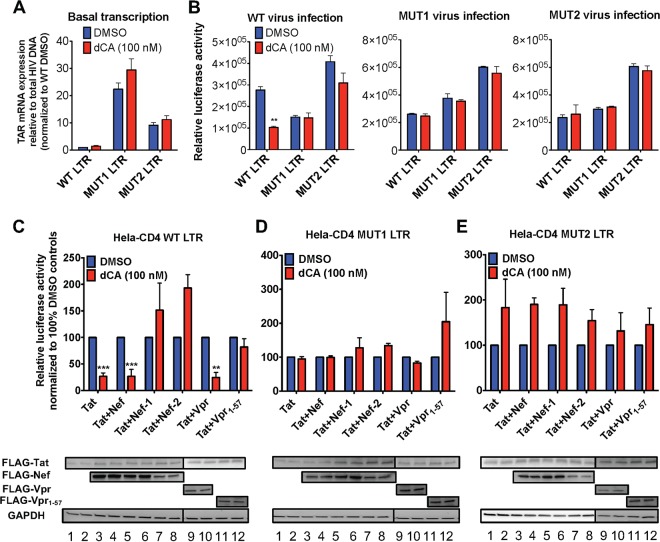
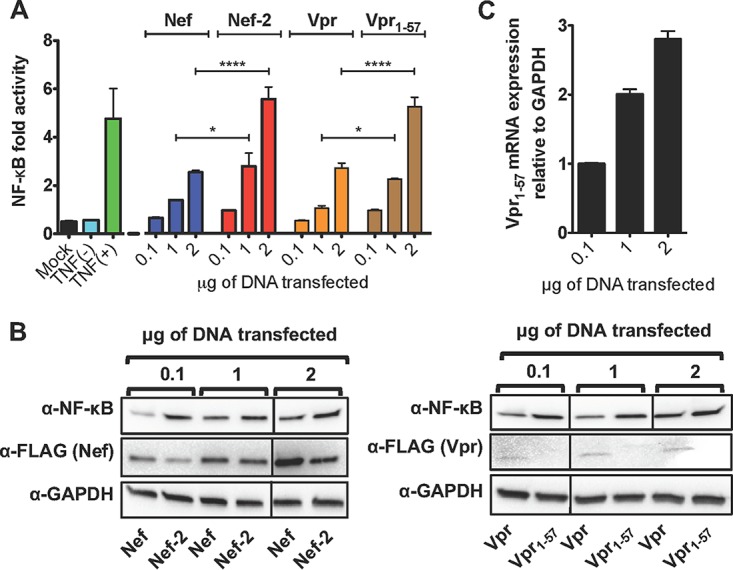
Human immunodeficiency virus type 1 (HIV-1) Tat binds the viral RNA structure transactivation-responsive element (TAR) and recruits transcriptional cofactors, amplifying viral mRNA expression. The Tat inhibitor didehydro-cortistatin A (dCA) promotes a state of persistent latency, refractory to viral reactivation. Here we investigated mechanisms of HIV-1 resistance to dCA in vitro Mutations in Tat and TAR were not identified, consistent with the high level of conservation of these elements. Instead, viruses resistant to dCA developed higher Tat-independent basal transcription. We identified a combination of mutations in the HIV-1 promoter that increased basal transcriptional activity and modifications in viral Nef and Vpr proteins that increased NF-κB activity. Importantly, these variants are unlikely to enter latency due to accrued transcriptional fitness and loss of sensitivity to Tat feedback loop regulation. Furthermore, cells infected with these variants become more susceptible to cytopathic effects and immune-mediated clearance. This is the first report of viral escape to a Tat inhibitor resulting in heightened Tat-independent activity, all while maintaining wild-type Tat and TAR.IMPORTANCE HIV-1 Tat enhances viral RNA transcription by binding to TAR and recruiting activating factors. Tat enhances its own transcription via a positive-feedback loop. Didehydro-cortistatin A (dCA) is a potent Tat inhibitor, reducing HIV-1 transcription and preventing viral rebound. dCA activity demonstrates the potential of the "block-and-lock" functional cure approaches. We investigated the viral genetic barrier to dCA resistance in vitro While mutations in Tat and TAR were not identified, mutations in the promoter and in the Nef and Vpr proteins promoted high Tat-independent activity. Promoter mutations increased the basal transcription, while Nef and Vpr mutations increased NF-κB nuclear translocation. This heightened transcriptional activity renders CD4+ T cells infected with these viruses more susceptible to cytotoxic T cell-mediated killing and to cell death by cytopathic effects. Results provide insights on drug resistance to a novel class of antiretrovirals and reveal novel aspects of viral transcriptional regulation.
Keywords: HIV promoter; HIV transcription; Nef; Tat inhibitor; Vpr; drug resistance; viral resistance.
Copyright © 2019 Mousseau et al.
Figures
Comment in
-
Unexpected Mutations in HIV-1 That Confer Resistance to the Tat Inhibitor Didehydro-Cortistatin A.mBio. 2019 Jul 9;10(4):e01547-19. doi: 10.1128/mBio.01547-19. mBio. 2019. PMID: 31289189 Free PMC article.
Similar articles
-
The Block-and-Lock Strategy for Human Immunodeficiency Virus Cure: Lessons Learned from Didehydro-Cortistatin A.J Infect Dis. 2021 Feb 15;223(12 Suppl 2):46-53. doi: 10.1093/infdis/jiaa681. J Infect Dis. 2021. PMID: 33586776 Free PMC article. Review.
-
The Tat Inhibitor Didehydro-Cortistatin A Prevents HIV-1 Reactivation from Latency.mBio. 2015 Jul 7;6(4):e00465. doi: 10.1128/mBio.00465-15. mBio. 2015. PMID: 26152583 Free PMC article.
-
Unexpected Mutations in HIV-1 That Confer Resistance to the Tat Inhibitor Didehydro-Cortistatin A.mBio. 2019 Jul 9;10(4):e01547-19. doi: 10.1128/mBio.01547-19. mBio. 2019. PMID: 31289189 Free PMC article.
-
Didehydro-Cortistatin A Inhibits HIV-1 by Specifically Binding to the Unstructured Basic Region of Tat.mBio. 2019 Feb 5;10(1):e02662-18. doi: 10.1128/mBio.02662-18. mBio. 2019. PMID: 30723126 Free PMC article.
-
Tat-Based Therapies as an Adjuvant for an HIV-1 Functional Cure.Viruses. 2020 Apr 8;12(4):415. doi: 10.3390/v12040415. Viruses. 2020. PMID: 32276443 Free PMC article. Review.
Cited by
-
Cure and Long-Term Remission Strategies.Methods Mol Biol. 2022;2407:391-428. doi: 10.1007/978-1-0716-1871-4_26. Methods Mol Biol. 2022. PMID: 34985678 Review.
-
Epigenetic crosstalk in chronic infection with HIV-1.Semin Immunopathol. 2020 Apr;42(2):187-200. doi: 10.1007/s00281-020-00783-3. Epub 2020 Feb 11. Semin Immunopathol. 2020. PMID: 32047948 Free PMC article. Review.
-
The Block-and-Lock Strategy for Human Immunodeficiency Virus Cure: Lessons Learned from Didehydro-Cortistatin A.J Infect Dis. 2021 Feb 15;223(12 Suppl 2):46-53. doi: 10.1093/infdis/jiaa681. J Infect Dis. 2021. PMID: 33586776 Free PMC article. Review.
-
Shock and kill, but don't miss the target.EBioMedicine. 2020 Aug;58:102906. doi: 10.1016/j.ebiom.2020.102906. Epub 2020 Jul 21. EBioMedicine. 2020. PMID: 32707449 Free PMC article. No abstract available.
-
Block and Lock HIV Cure Strategies to Control the Latent Reservoir.Front Cell Infect Microbiol. 2020 Aug 14;10:424. doi: 10.3389/fcimb.2020.00424. eCollection 2020. Front Cell Infect Microbiol. 2020. PMID: 32923412 Free PMC article. Review.
References
-
- Condra JH, Schleif WA, Blahy OM, Gabryelski LJ, Graham DJ, Quintero JC, Rhodes A, Robbins HL, Roth E, Shivaprakash M, Titus D, Yang T, Tepplert H, Squires KE, Deutsch PJ, Emini EA. 1995. In vivo emergence of HIV-1 variants resistant to multiple protease inhibitors. Nature 374:569–571. doi:10.1038/374569a0. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
