

Human 5' UTR design and variant effect prediction from a massively parallel translation assay
- PMID: 31267113
- PMCID: PMC7100133
- DOI: 10.1038/s41587-019-0164-5
Human 5' UTR design and variant effect prediction from a massively parallel translation assay
Abstract
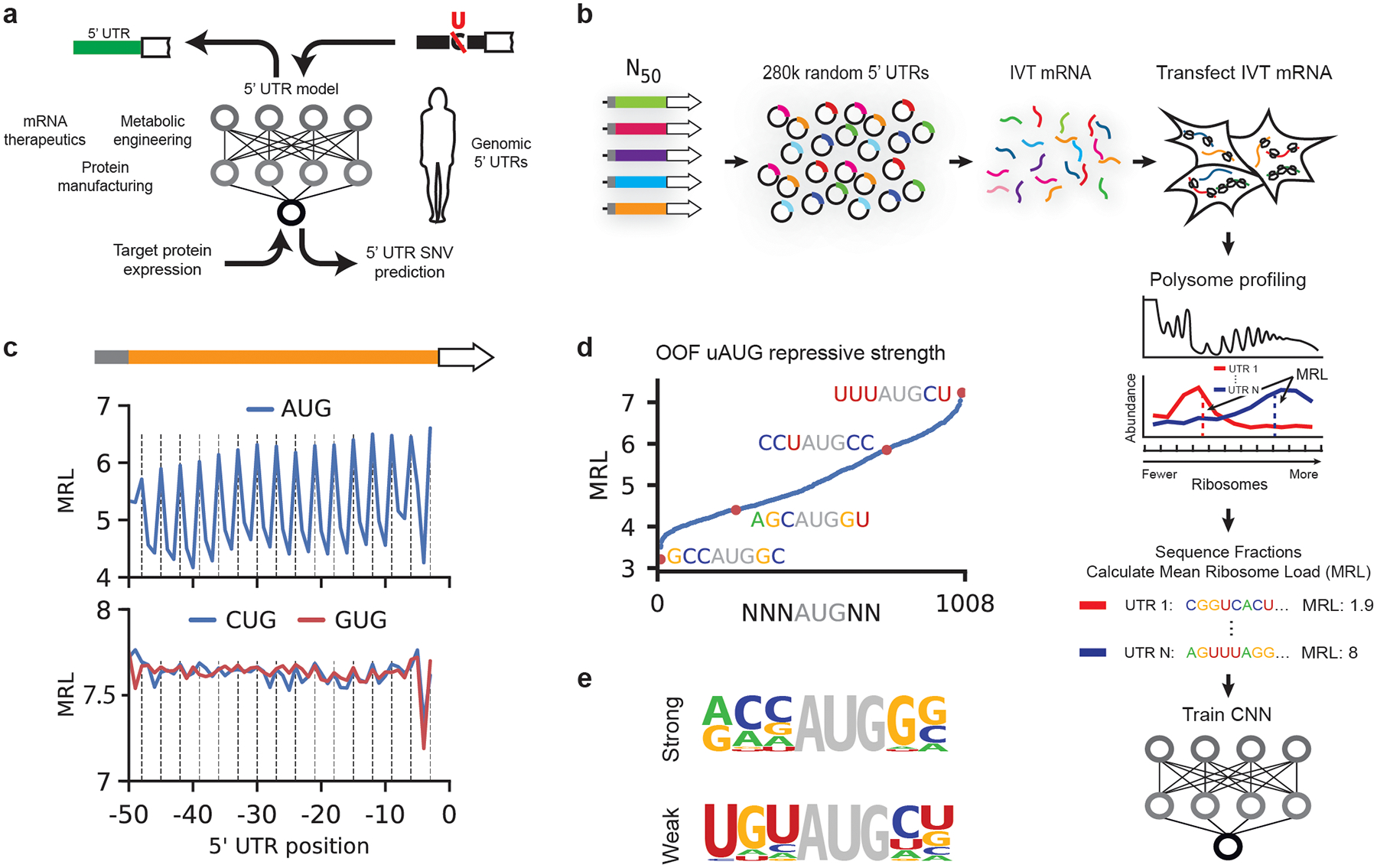
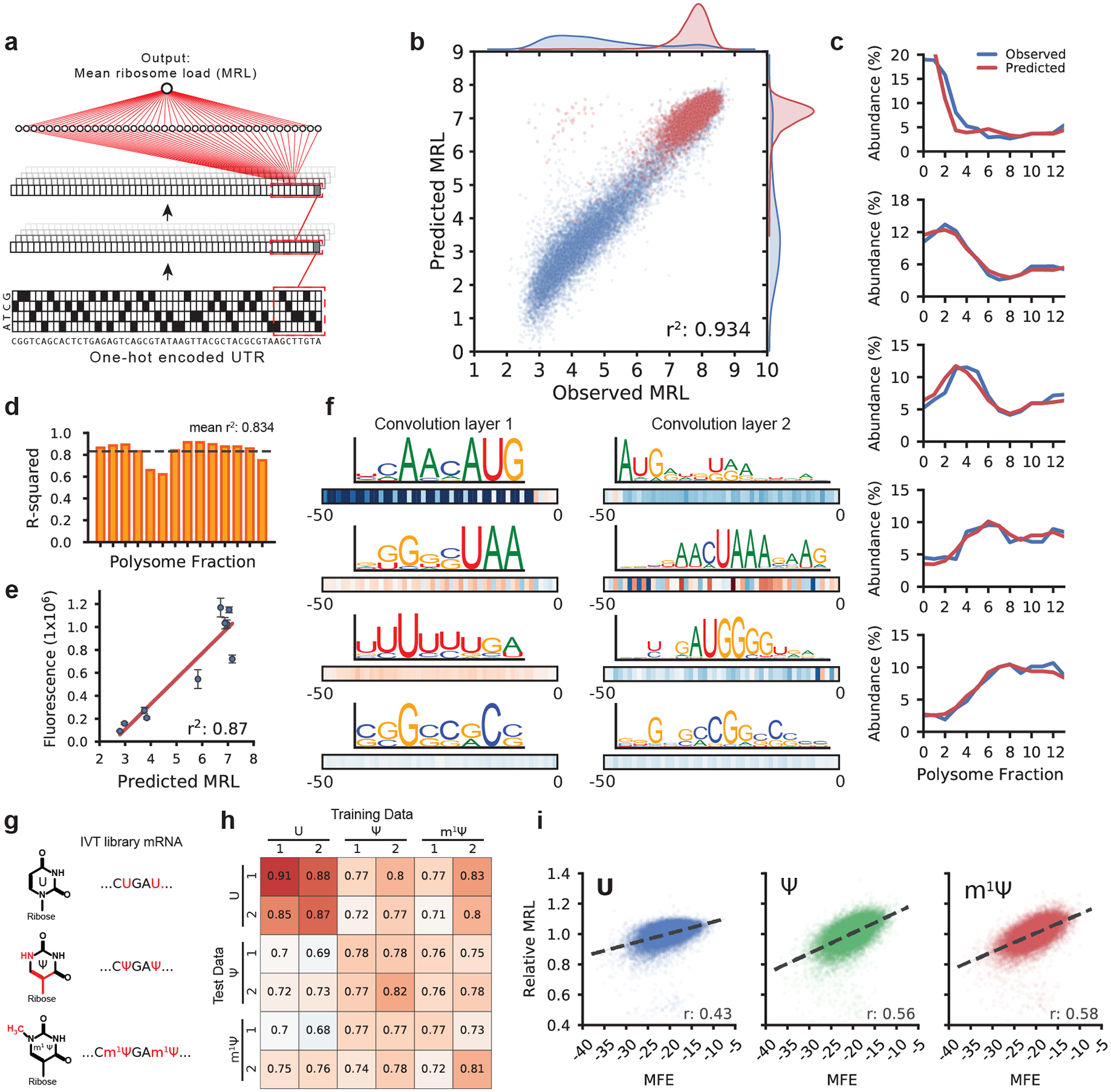
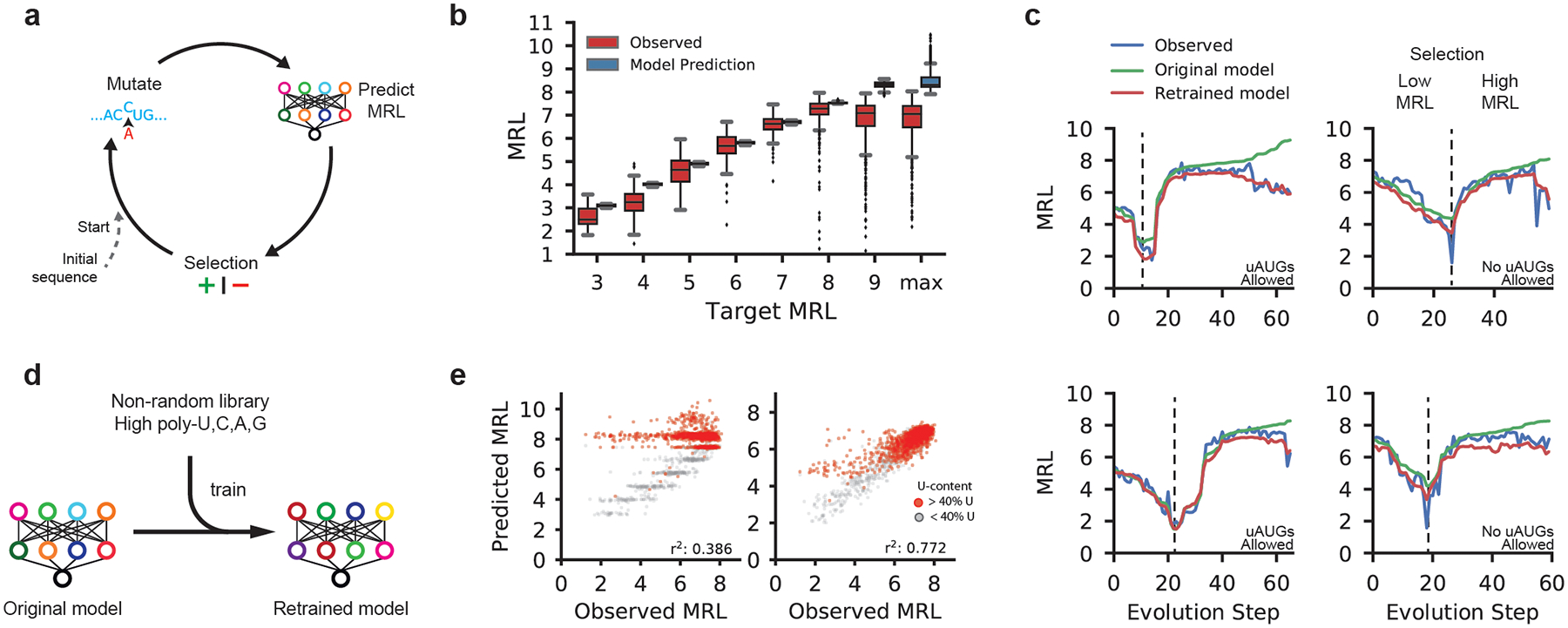
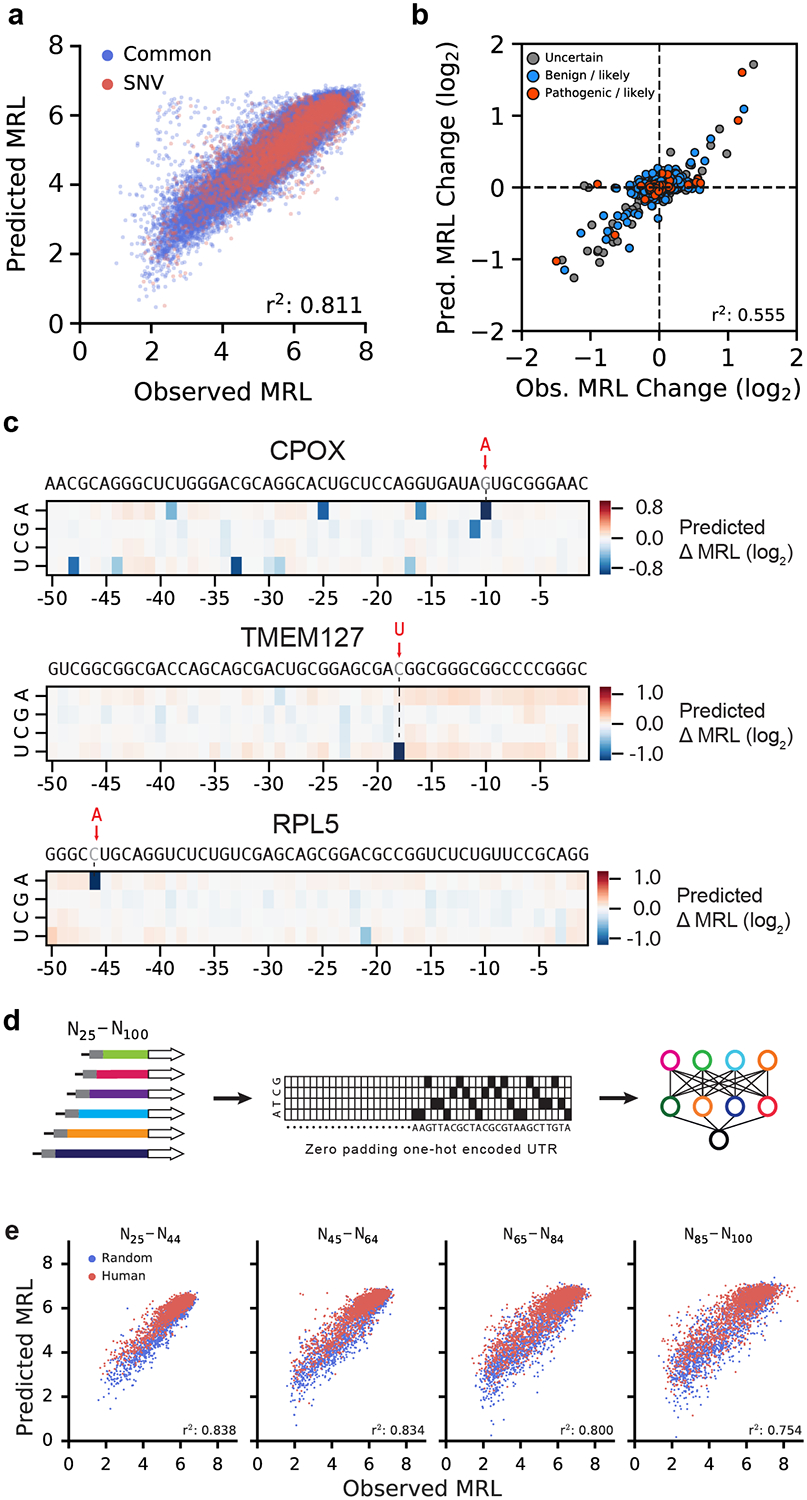
The ability to predict the impact of cis-regulatory sequences on gene expression would facilitate discovery in fundamental and applied biology. Here we combine polysome profiling of a library of 280,000 randomized 5' untranslated regions (UTRs) with deep learning to build a predictive model that relates human 5' UTR sequence to translation. Together with a genetic algorithm, we use the model to engineer new 5' UTRs that accurately direct specified levels of ribosome loading, providing the ability to tune sequences for optimal protein expression. We show that the same approach can be extended to chemically modified RNA, an important feature for applications in mRNA therapeutics and synthetic biology. We test 35,212 truncated human 5' UTRs and 3,577 naturally occurring variants and show that the model predicts ribosome loading of these sequences. Finally, we provide evidence of 45 single-nucleotide variants (SNVs) associated with human diseases that substantially change ribosome loading and thus may represent a molecular basis for disease.
Conflict of interest statement
Competing interests
PJS, BW, GS, and DRM declare no competing interests. DR, VP, and IM are employees and shareholders of Moderna Therapeutics.
Figures
References
Methods-only References
-
- Martin M Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal (2011). doi:10.14806/ej.17.1.200 - DOI
-
- Chollet F Keras (2015). URL http://keras.io (2017).
-
- Abadi M et al. TensorFlow : A System for Large-Scale Machine Learning This paper is included in the Proceedings of the TensorFlow : A system for large-scale machine learning. Proc 12th USENIX Conf. Oper. Syst. Des. Implement (2016). doi:10.1126/science.aab4113.4 - DOI
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
