

An improved method for circular RNA purification using RNase R that efficiently removes linear RNAs containing G-quadruplexes or structured 3' ends
- PMID: 31269210
- PMCID: PMC6895279
- DOI: 10.1093/nar/gkz576
An improved method for circular RNA purification using RNase R that efficiently removes linear RNAs containing G-quadruplexes or structured 3' ends
Abstract
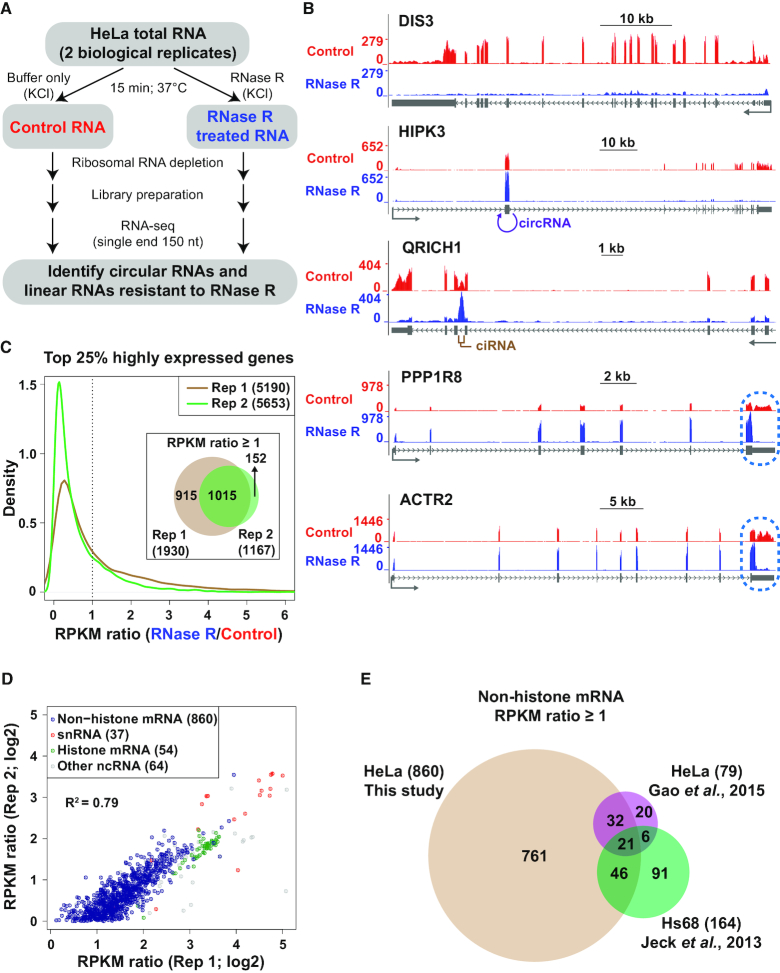
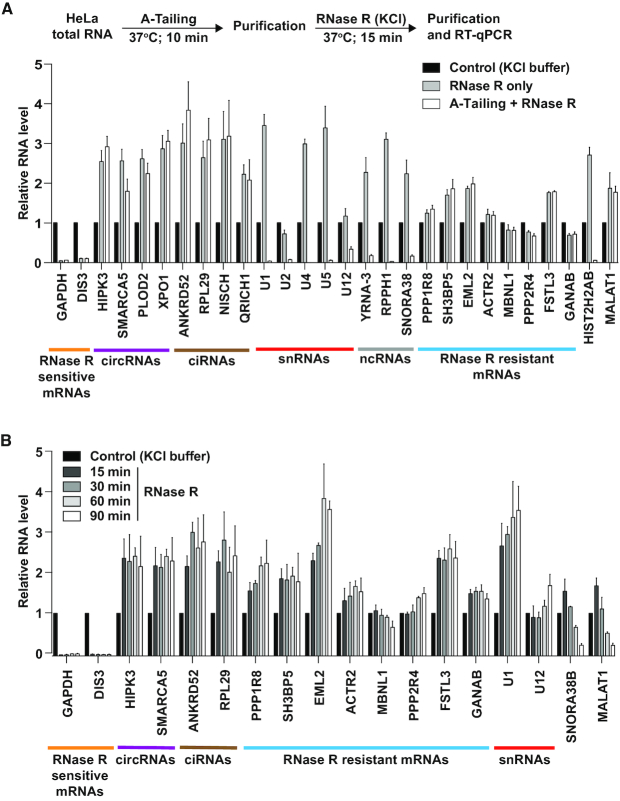
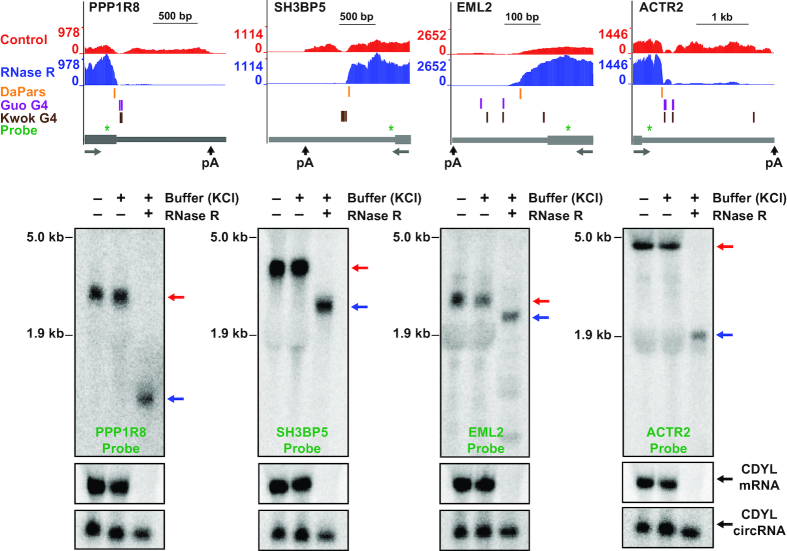
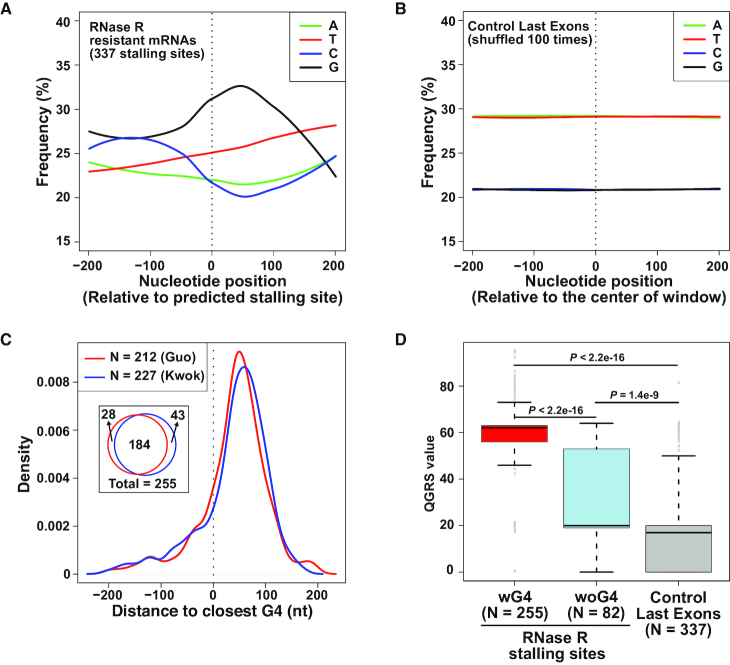
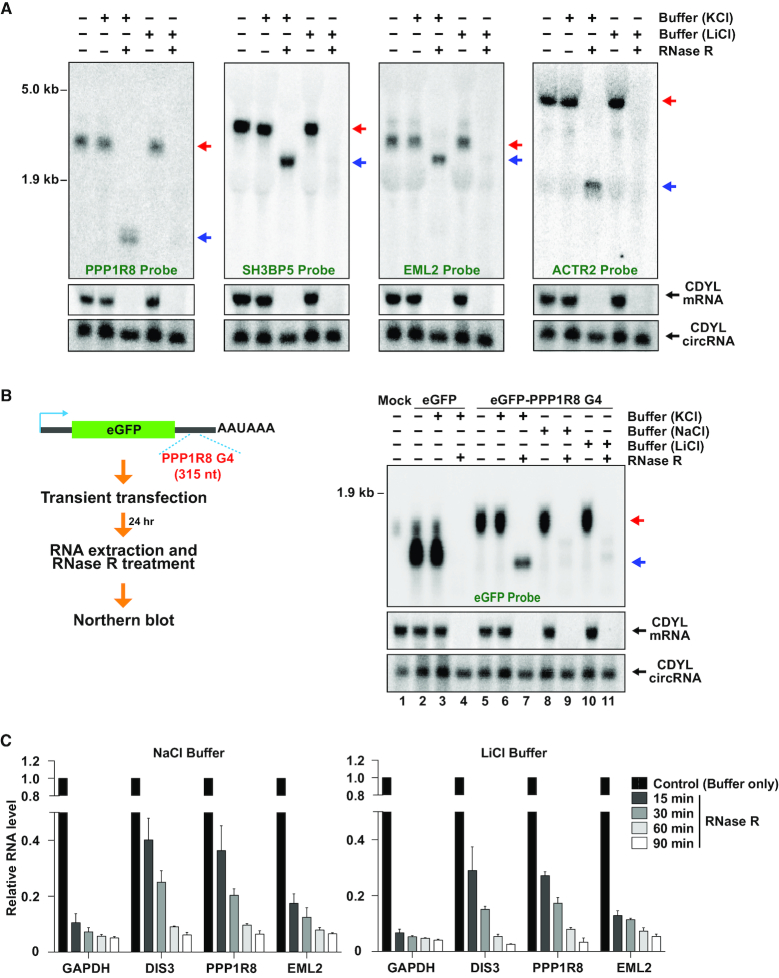
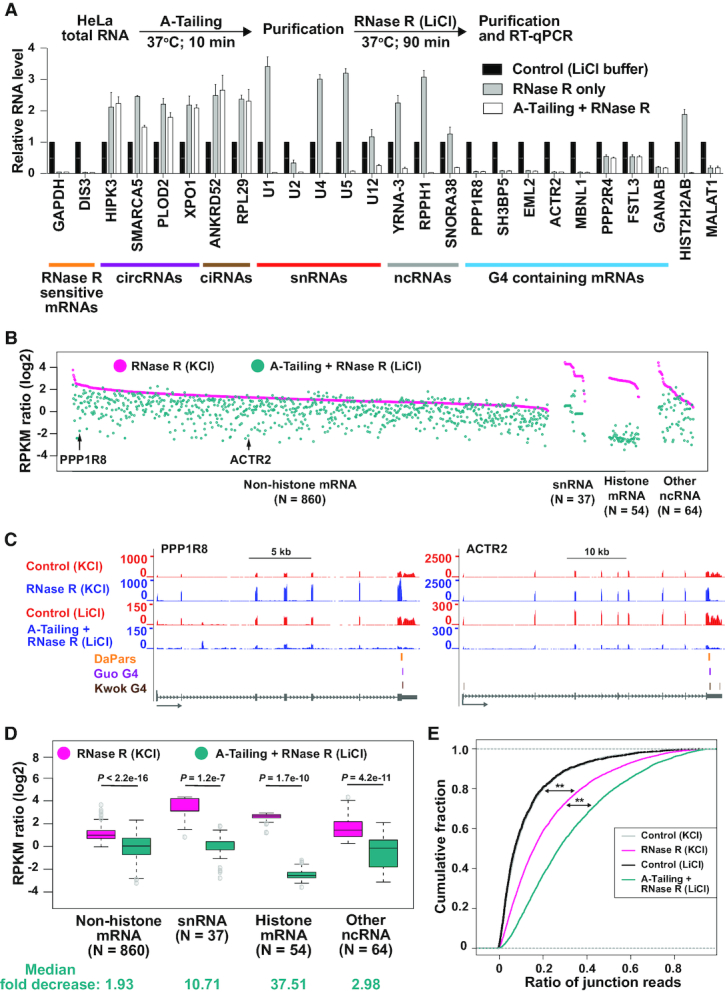
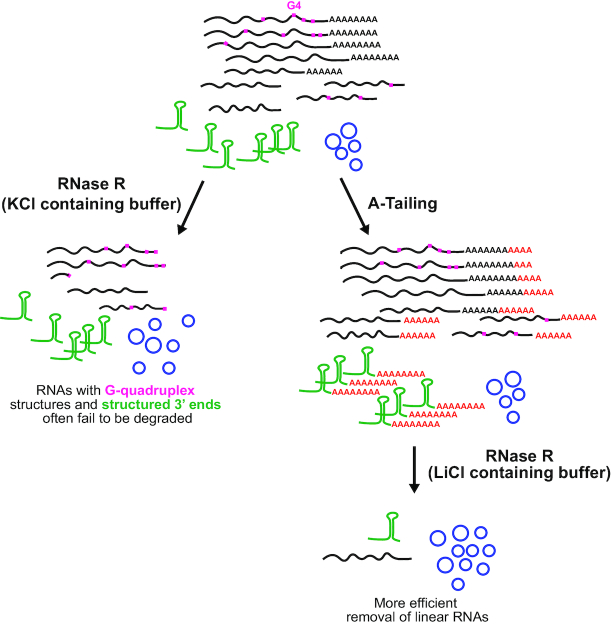
Thousands of eukaryotic protein-coding genes generate circular RNAs that have covalently linked ends and are resistant to degradation by exonucleases. To prove their circularity as well as biochemically enrich these transcripts, it has become standard in the field to use the 3'-5' exonuclease RNase R. Here, we demonstrate that standard protocols involving RNase R can fail to digest >20% of all highly expressed linear RNAs, but these shortcomings can largely be overcome. RNAs with highly structured 3' ends, including snRNAs and histone mRNAs, are naturally resistant to RNase R, but can be efficiently degraded once a poly(A) tail has been added to their ends. In addition, RNase R stalls in the body of many polyadenylated mRNAs, especially at G-rich sequences that have been previously annotated as G-quadruplex (G4) structures. Upon replacing K+ (which stabilizes G4s) with Li+ in the reaction buffer, we find that RNase R is now able to proceed through these sequences and fully degrade the mRNAs in their entirety. In total, our results provide important improvements to the current methods used to isolate circular RNAs as well as a way to reveal RNA structures that may naturally inhibit degradation by cellular exonucleases.
© The Author(s) 2019. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
