

In Vivo ERK1/2 Reporter Predictively Models Response and Resistance to Combined BRAF and MEK Inhibitors in Melanoma
- PMID: 31270153
- PMCID: PMC6726573
- DOI: 10.1158/1535-7163.MCT-18-1056
In Vivo ERK1/2 Reporter Predictively Models Response and Resistance to Combined BRAF and MEK Inhibitors in Melanoma
Abstract
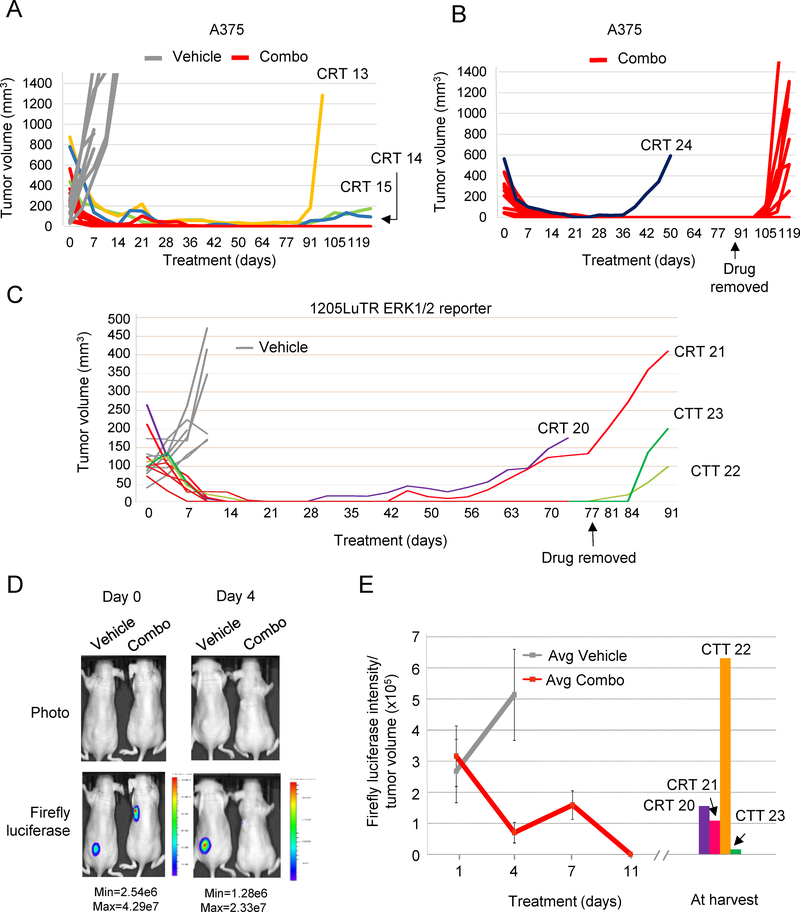
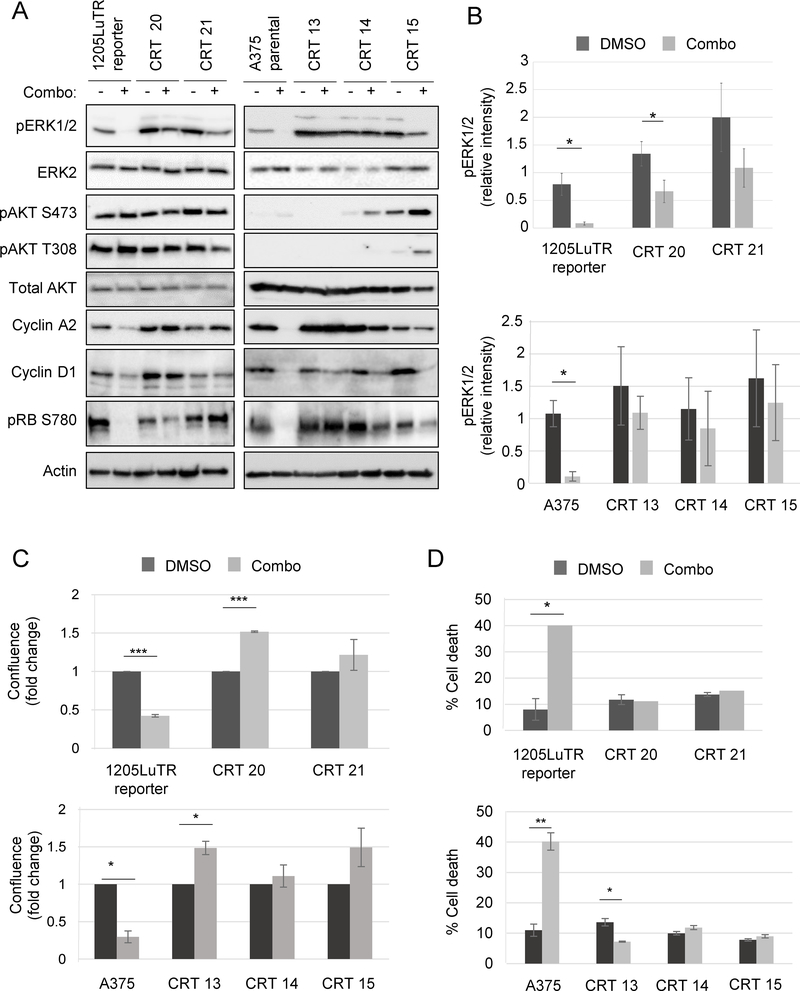
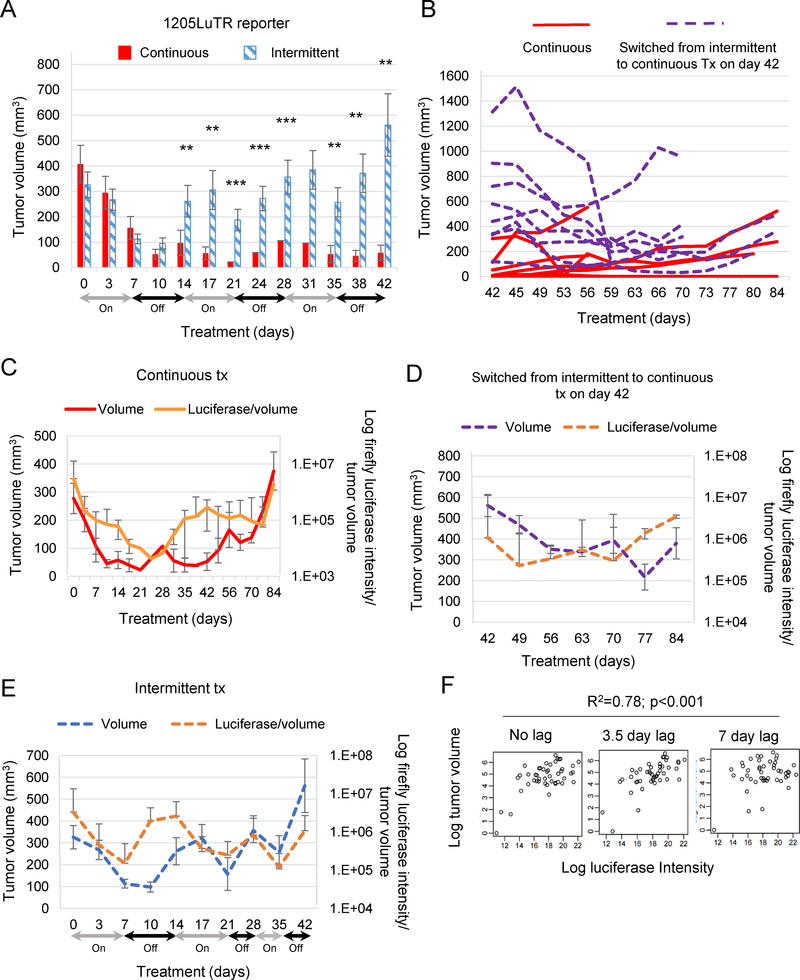
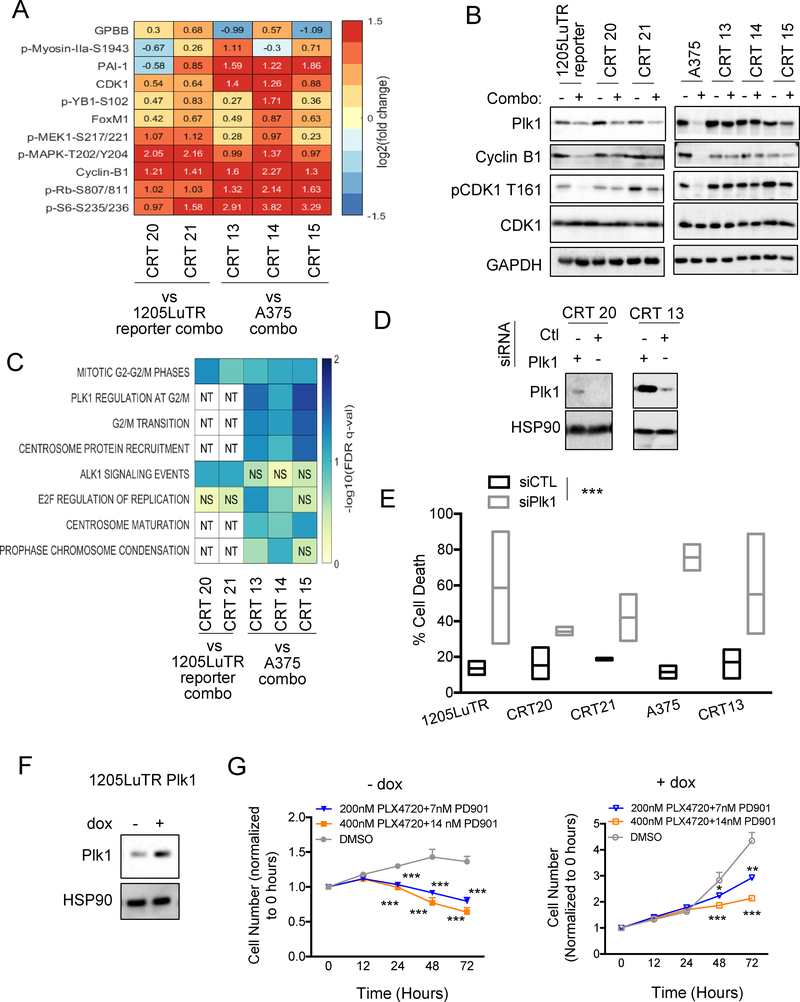
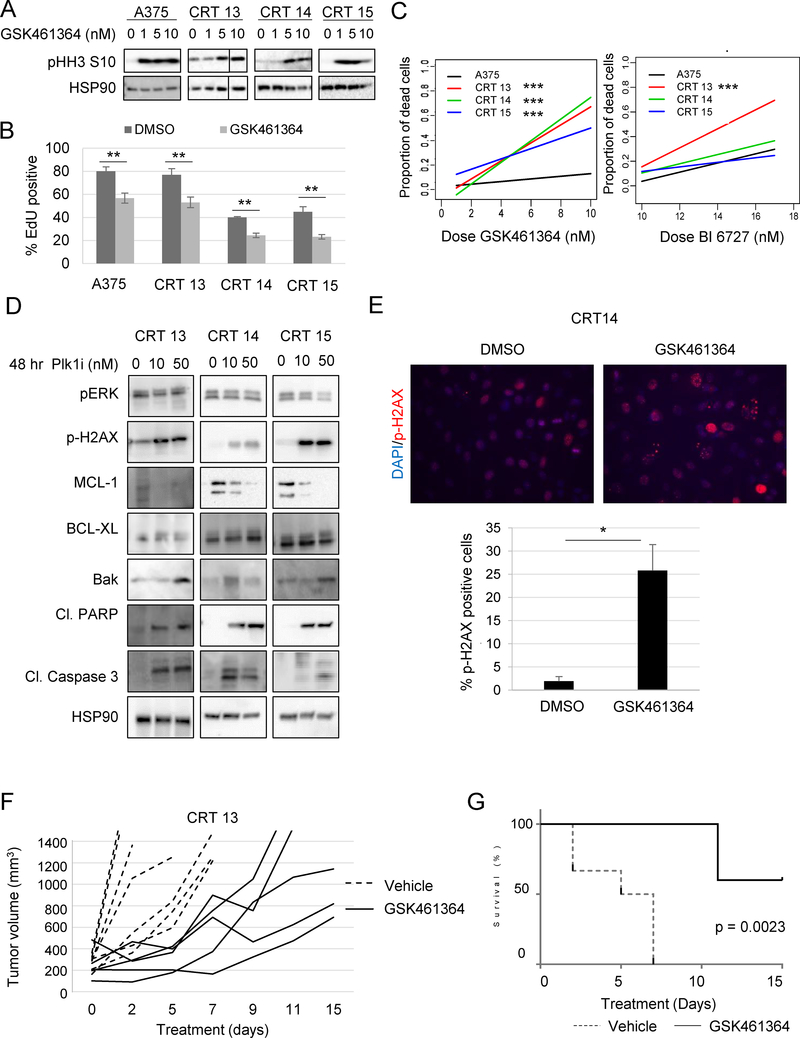
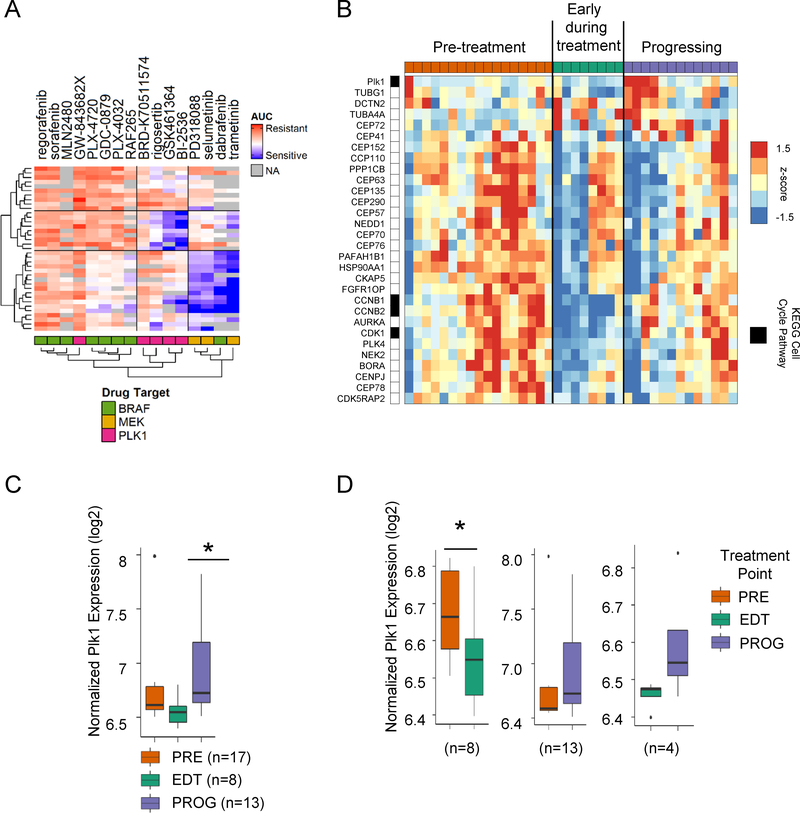
Combined BRAF and MEK inhibition is a standard of care in patients with advanced BRAF-mutant melanoma, but acquired resistance remains a challenge that limits response durability. Here, we quantitated in vivo ERK1/2 activity and tumor response associated with resistance to combined BRAF and MEK inhibition in mutant BRAF xenografts. We found that ERK1/2 pathway reactivation preceded the growth of resistant tumors. Moreover, we detected a subset of cells that not only persisted throughout long-term treatment but restored ERK1/2 signaling and grew upon drug removal. Cell lines derived from combination-resistant tumors (CRT) exhibited elevated ERK1/2 phosphorylation, which were sensitive to ERK1/2 inhibition. In some CRTs, we detected a tandem duplication of the BRAF kinase domain. Monitoring ERK1/2 activity in vivo was efficacious in predicting tumor response during intermittent treatment. We observed maintained expression of the mitotic regulator, polo-like kinase 1 (Plk1), in melanoma resistant to BRAF and MEK inhibitors. Plk1 inhibition induced apoptosis in CRTs, leading to slowed growth of BRAF and MEK inhibitor-resistant tumors in vivo These data demonstrate the utility of in vivo ERK1/2 pathway reporting as a tool to optimize clinical dosing schemes and establish suppression of Plk1 as potential salvage therapy for BRAF inhibitor and MEK inhibitor-resistant melanoma.
©2019 American Association for Cancer Research.
Figures
References
-
- Conner SR, Scott G, Aplin AE. Adhesion-dependent activation of the ERK½ cascade is by-passed in melanoma cells. J Biol Chem 2003;278:34548–54. - PubMed
-
- Hingorani SR, Jacobetz MA, Robertson GP, Herlyn M, Tuveson DA. Suppression of BRAF(V599E) in human melanoma abrogates transformation. Cancer research 2003;63:5198–202. - PubMed
-
- Flaherty KT, Robert C, Hersey P, Nathan P, Garbe C, Milhem M, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. The New England journal of medicine 2012;367:107–14. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
