

DNA assembly for nanopore data storage readout
- PMID: 31270330
- PMCID: PMC6610119
- DOI: 10.1038/s41467-019-10978-4
DNA assembly for nanopore data storage readout
Abstract
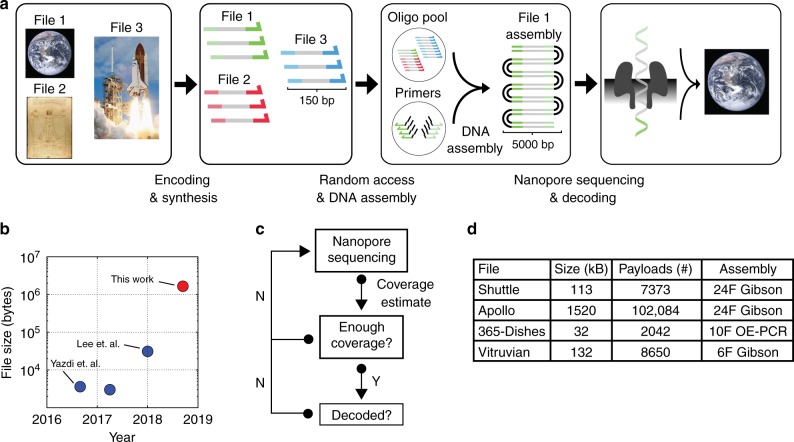
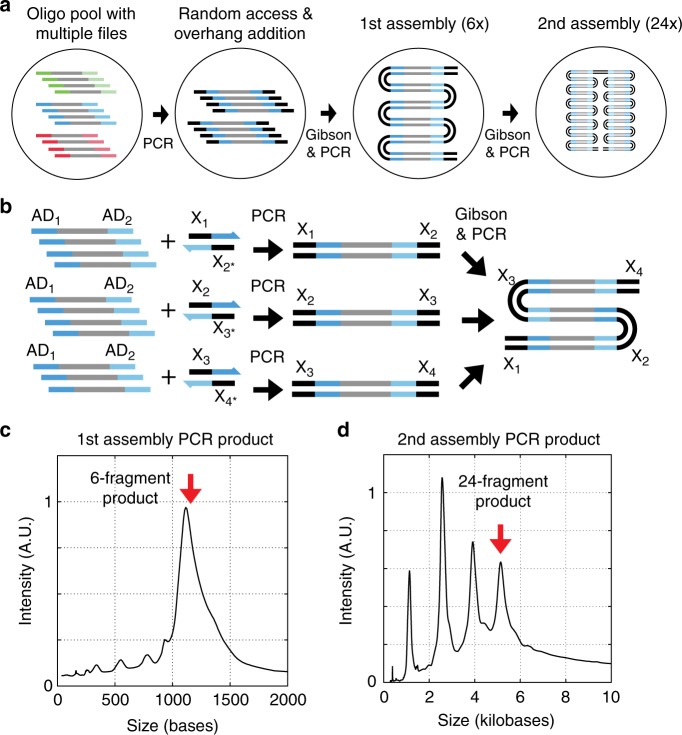
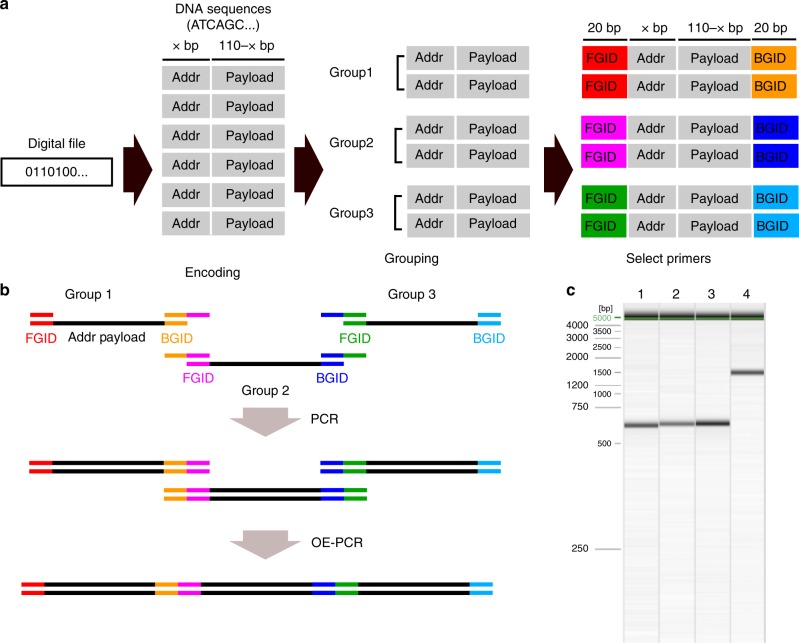
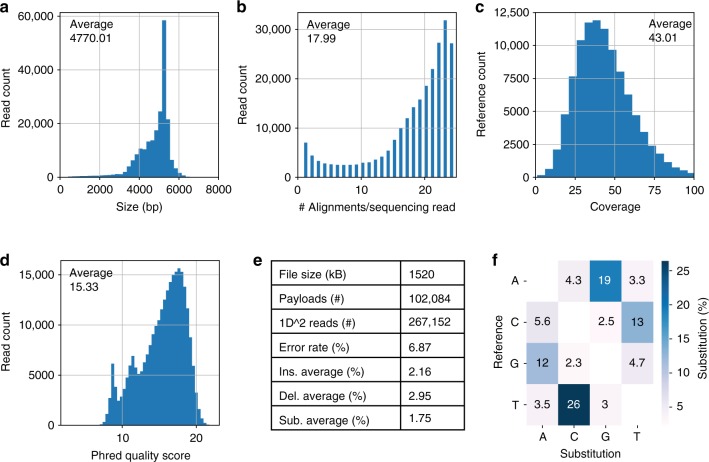
Synthetic DNA is becoming an attractive substrate for digital data storage due to its density, durability, and relevance in biological research. A major challenge in making DNA data storage a reality is that reading DNA back into data using sequencing by synthesis remains a laborious, slow and expensive process. Here, we demonstrate successful decoding of 1.67 megabytes of information stored in short fragments of synthetic DNA using a portable nanopore sequencing platform. We design and validate an assembly strategy for DNA storage that drastically increases the throughput of nanopore sequencing. Importantly, this assembly strategy is generalizable to any application that requires nanopore sequencing of small DNA amplicons.
Conflict of interest statement
Y-J. C., S.D.A., and K.S. are Microsoft employees. The remaining authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
