

Somatic mutations and cell identity linked by Genotyping of Transcriptomes
- PMID: 31270458
- PMCID: PMC6782071
- DOI: 10.1038/s41586-019-1367-0
Somatic mutations and cell identity linked by Genotyping of Transcriptomes
Abstract
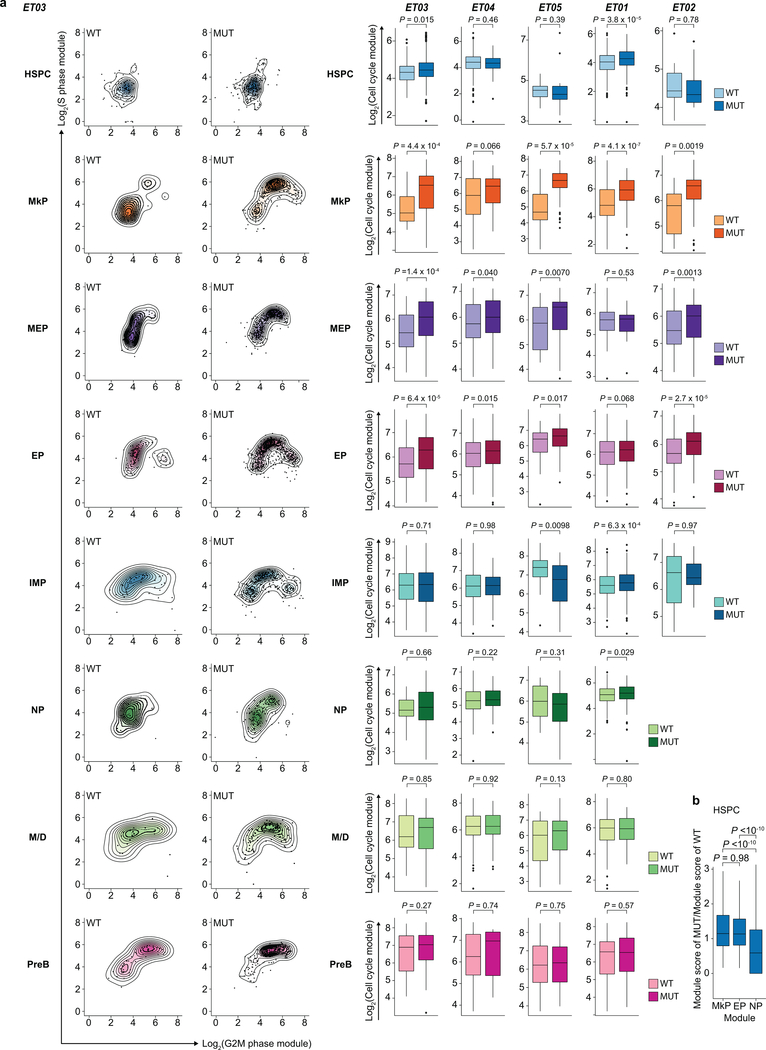
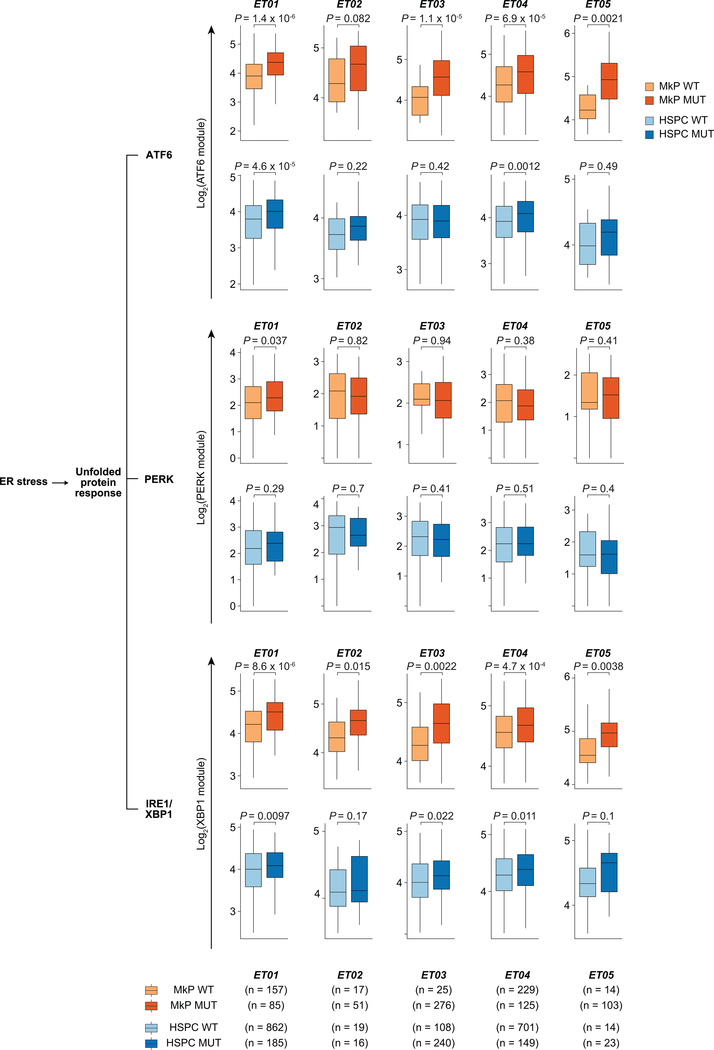
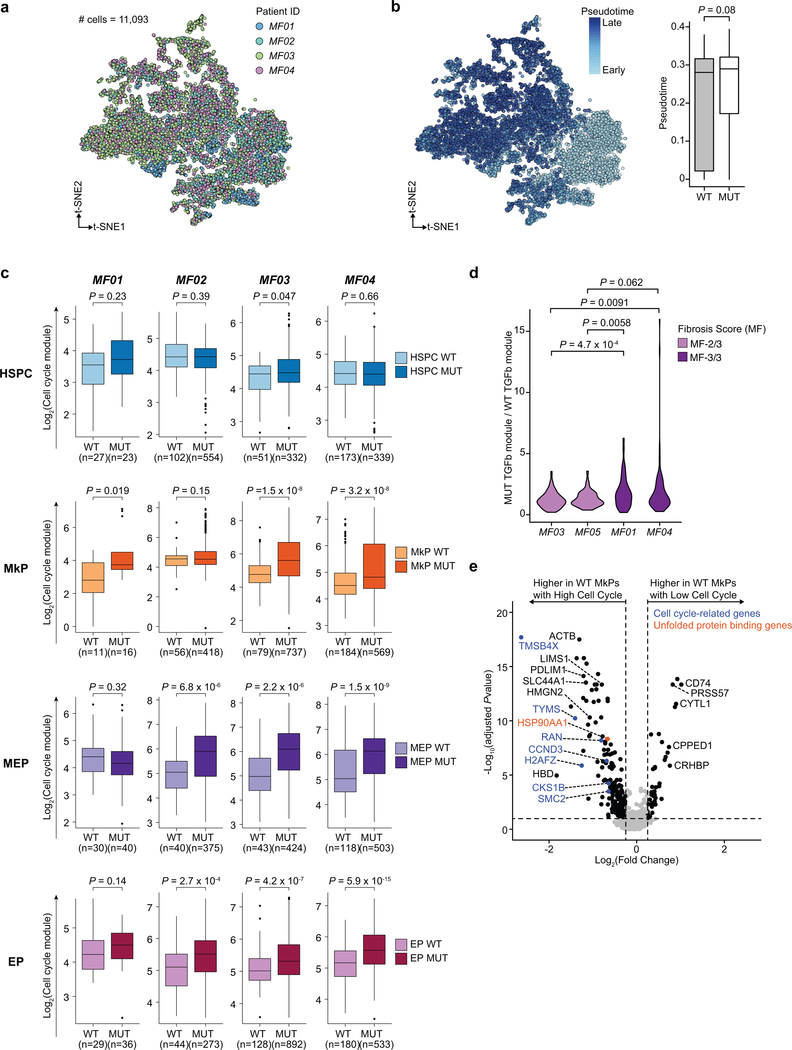
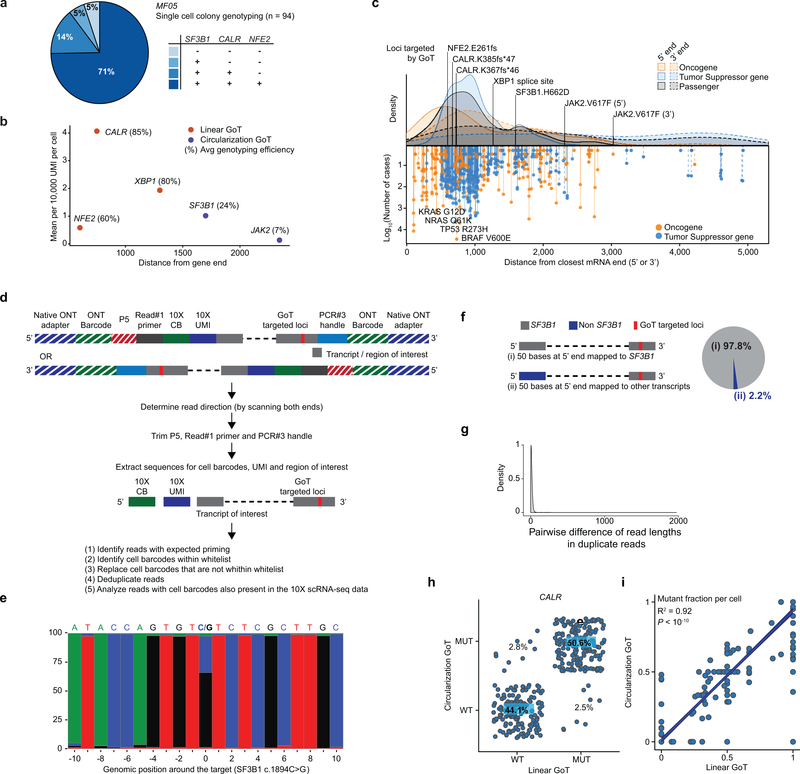
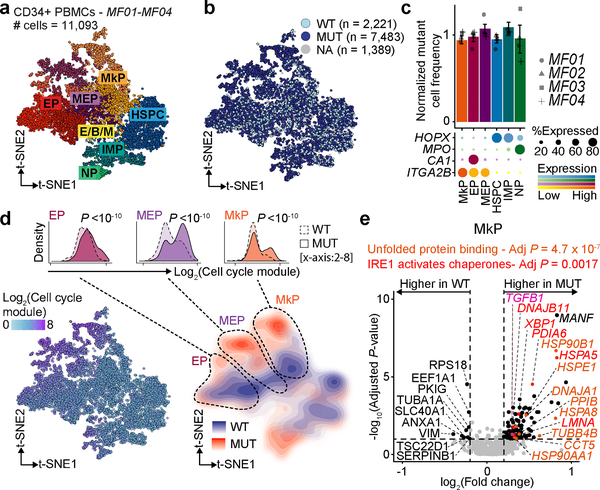
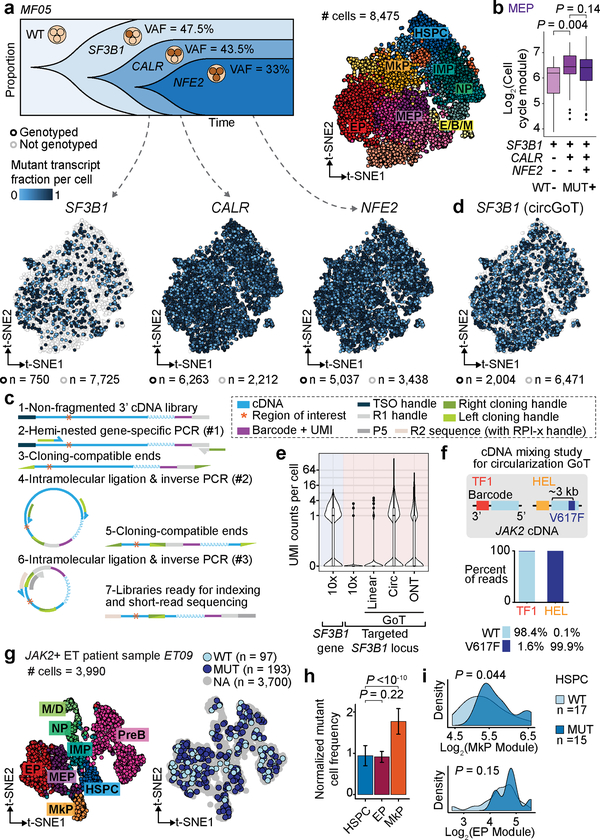
Defining the transcriptomic identity of malignant cells is challenging in the absence of surface markers that distinguish cancer clones from one another, or from admixed non-neoplastic cells. To address this challenge, here we developed Genotyping of Transcriptomes (GoT), a method to integrate genotyping with high-throughput droplet-based single-cell RNA sequencing. We apply GoT to profile 38,290 CD34+ cells from patients with CALR-mutated myeloproliferative neoplasms to study how somatic mutations corrupt the complex process of human haematopoiesis. High-resolution mapping of malignant versus normal haematopoietic progenitors revealed an increasing fitness advantage with myeloid differentiation of cells with mutated CALR. We identified the unfolded protein response as a predominant outcome of CALR mutations, with a considerable dependency on cell identity, as well as upregulation of the NF-κB pathway specifically in uncommitted stem cells. We further extended the GoT toolkit to genotype multiple targets and loci that are distant from transcript ends. Together, these findings reveal that the transcriptional output of somatic mutations in myeloproliferative neoplasms is dependent on the native cell identity.
Conflict of interest statement
Figures
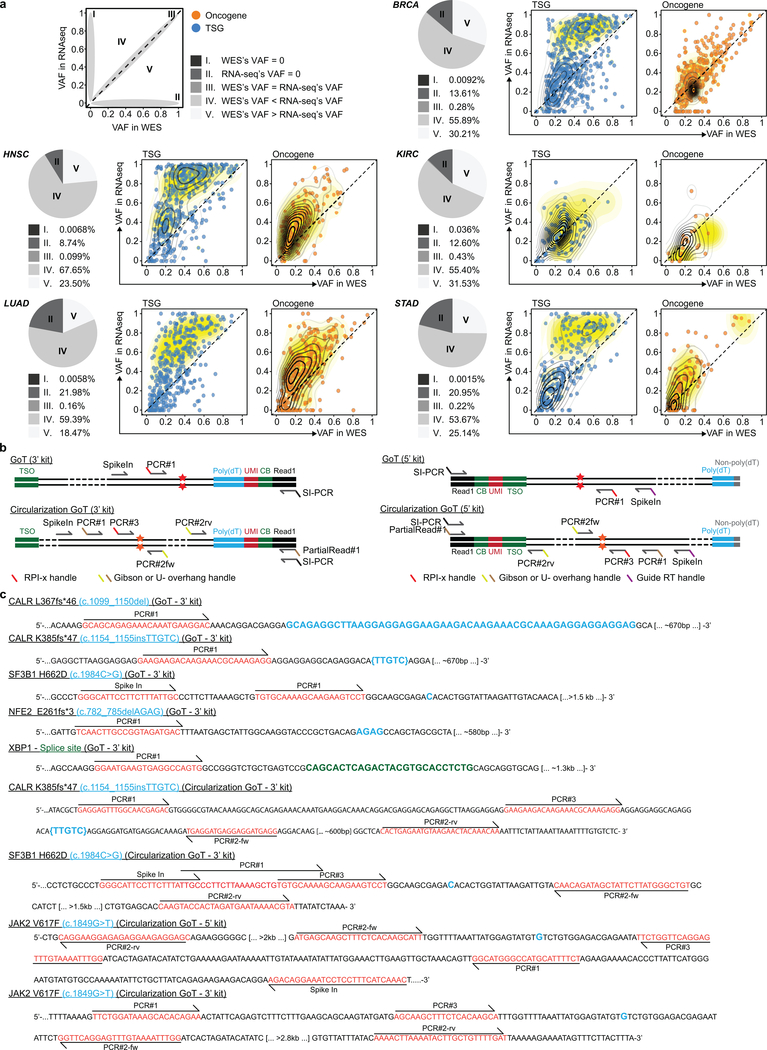
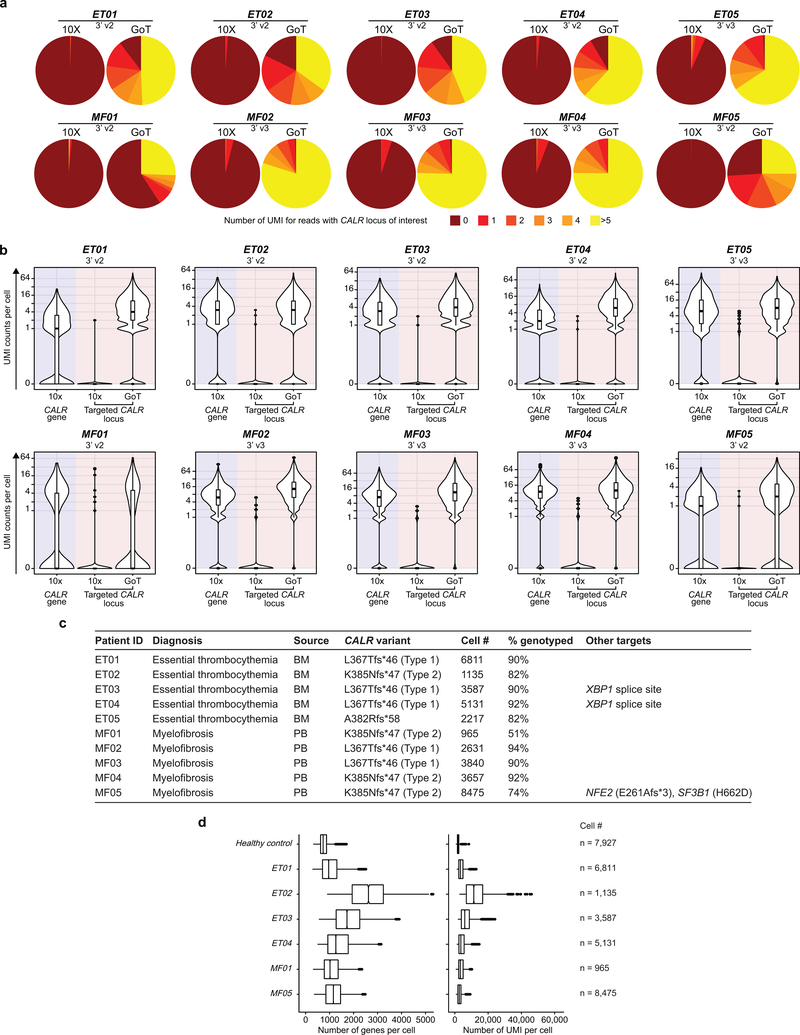
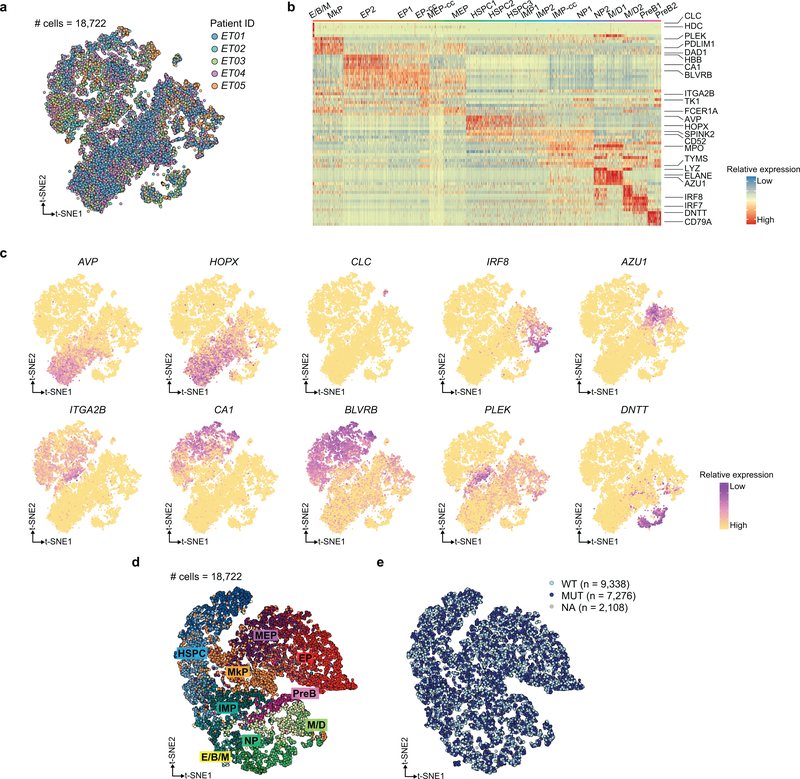
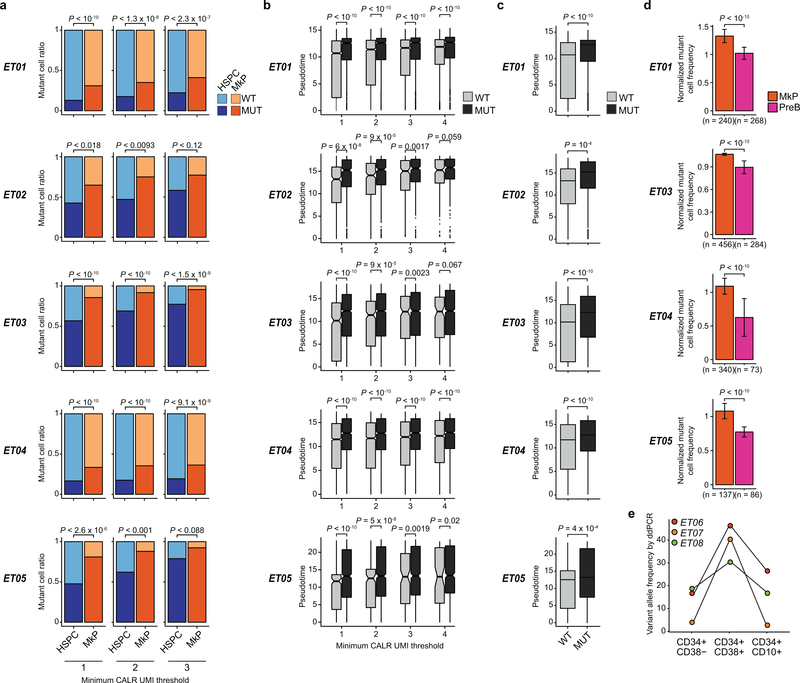
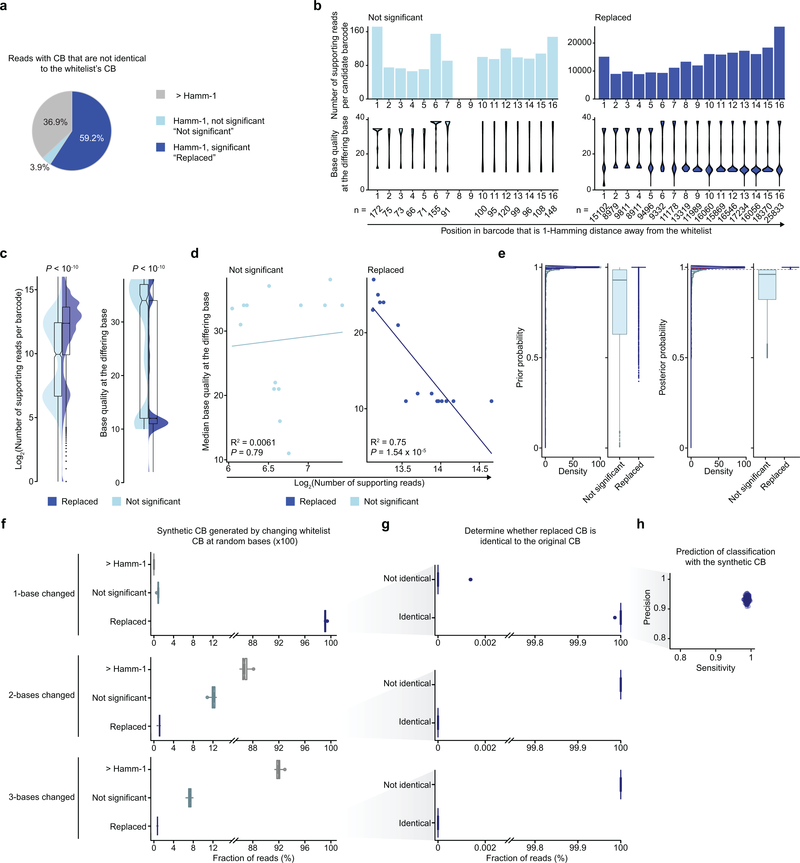
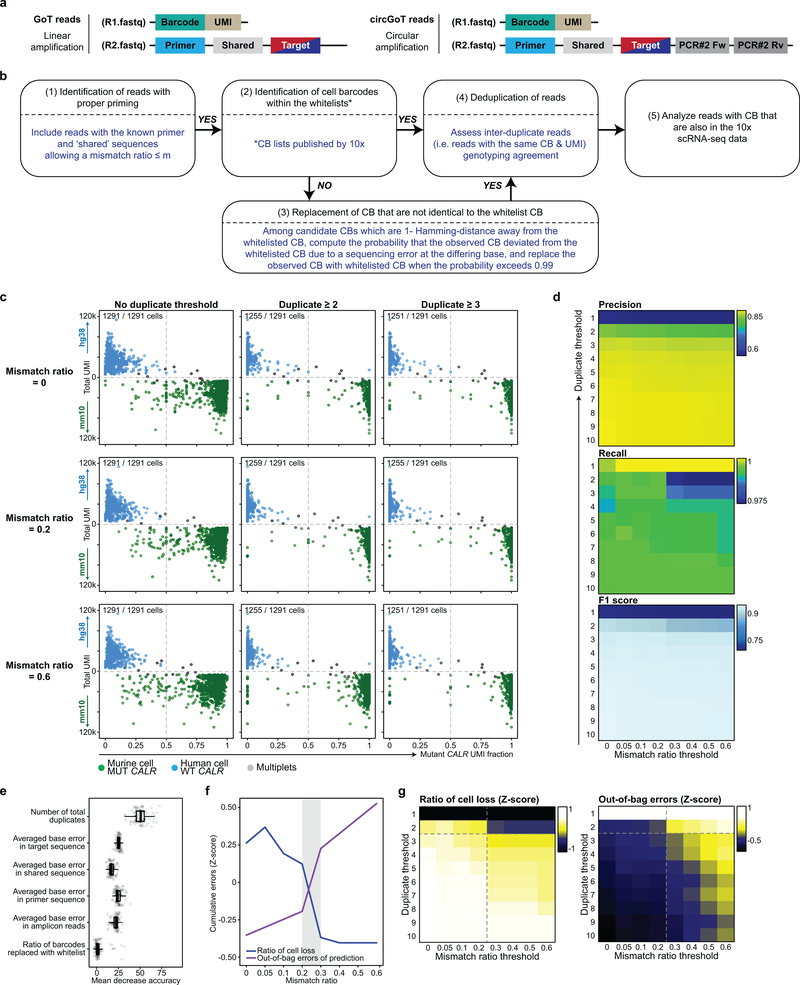
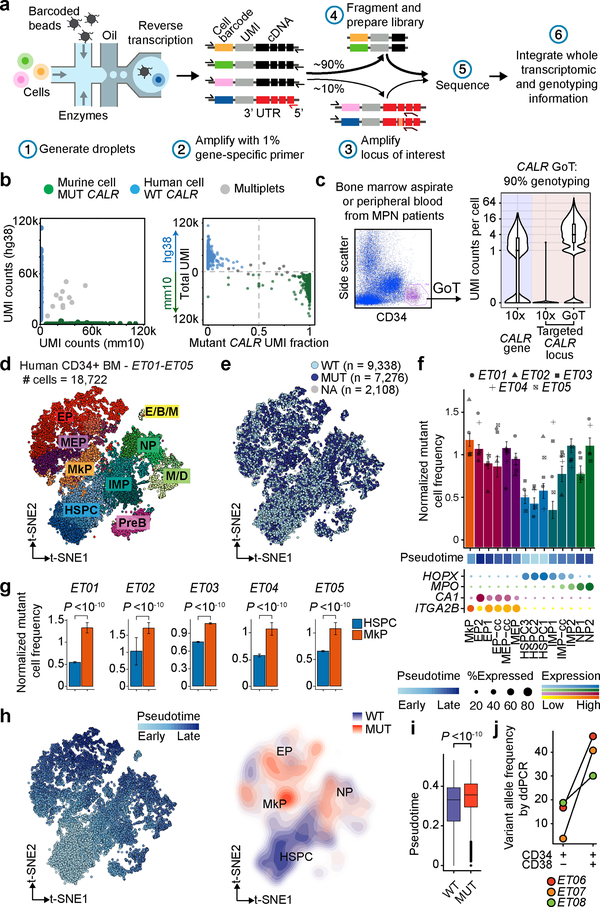

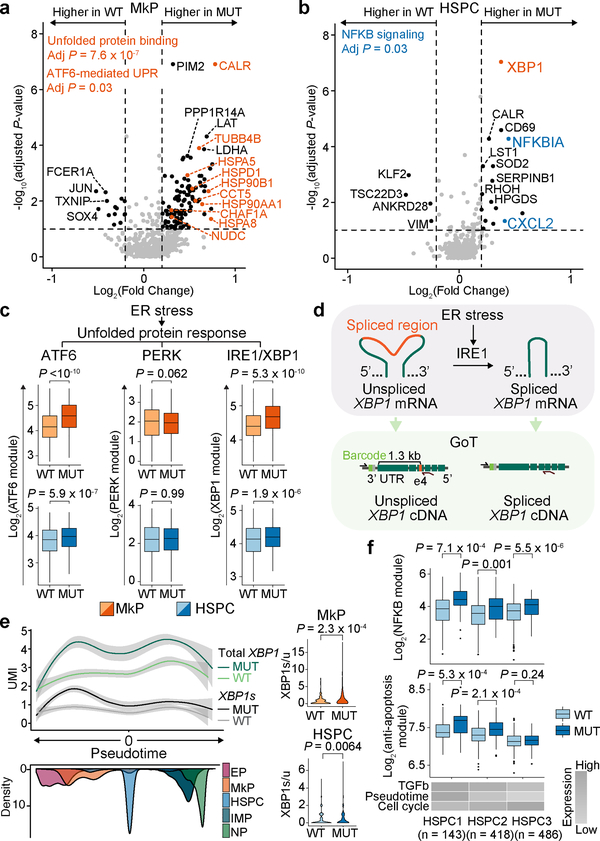
Comment in
-
How mutations express themselves in blood-cell production.Nature. 2019 Jul;571(7765):329-330. doi: 10.1038/d41586-019-02028-2. Nature. 2019. PMID: 31308526 No abstract available.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
