

Pooled clone collections by multiplexed CRISPR-Cas12a-assisted gene tagging in yeast
- PMID: 31273196
- PMCID: PMC6609715
- DOI: 10.1038/s41467-019-10816-7
Pooled clone collections by multiplexed CRISPR-Cas12a-assisted gene tagging in yeast
Abstract
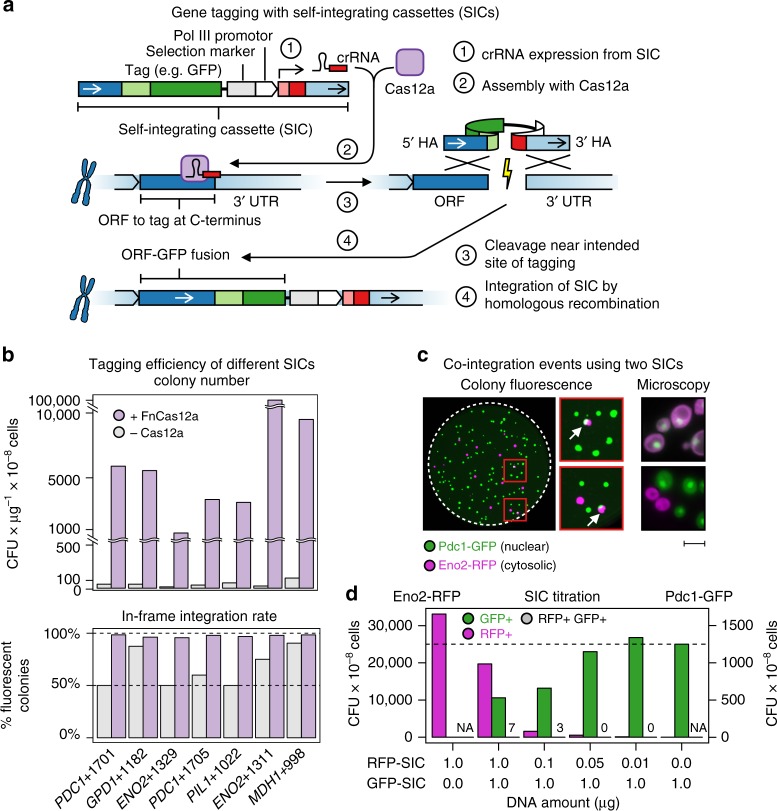
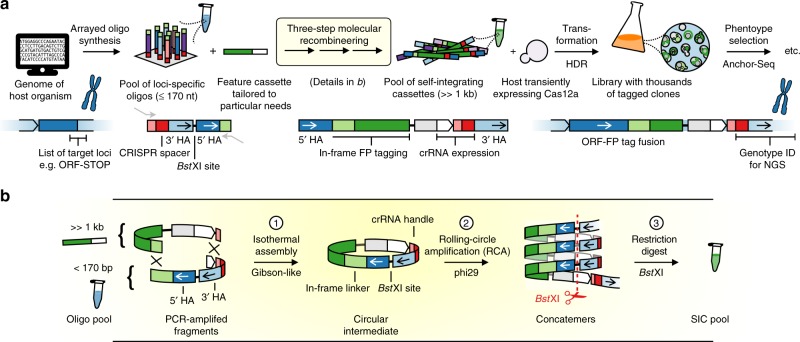
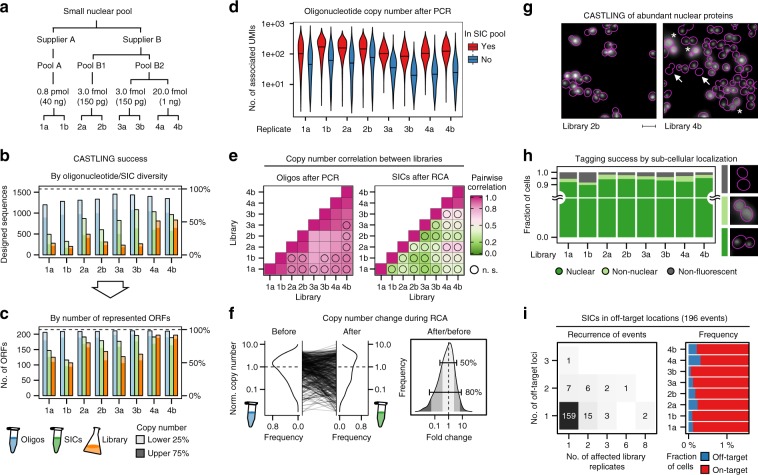
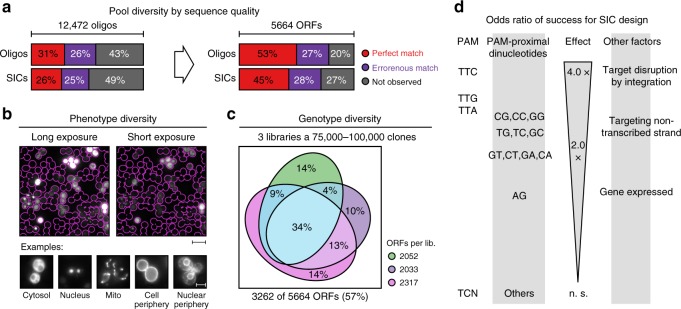
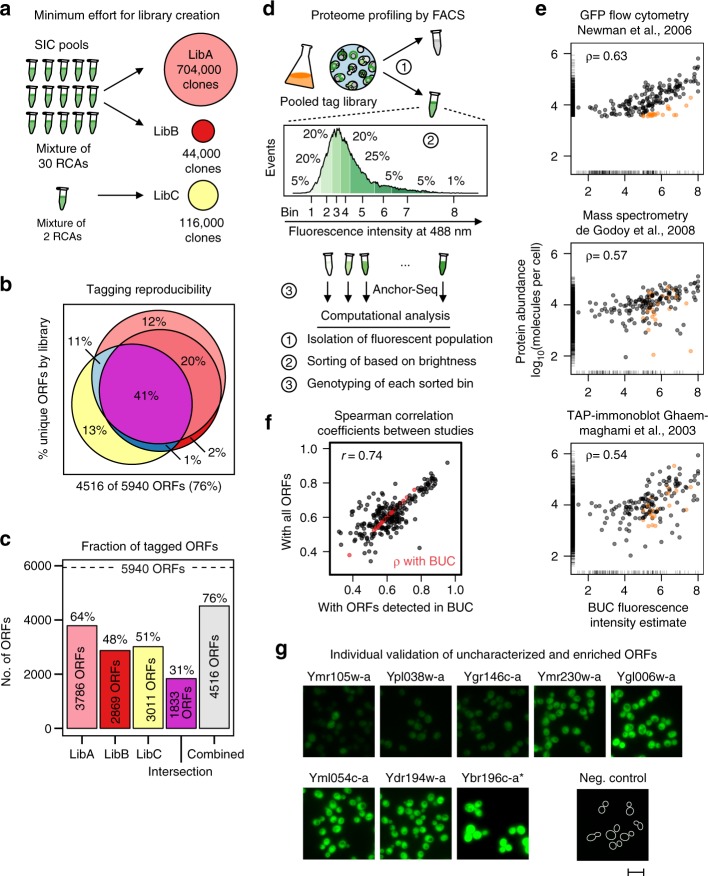
Clone collections of modified strains ("libraries") are a major resource for systematic studies with the yeast Saccharomyces cerevisiae. Construction of such libraries is time-consuming, costly and confined to the genetic background of a specific yeast strain. To overcome these limitations, we present CRISPR-Cas12a (Cpf1)-assisted tag library engineering (CASTLING) for multiplexed strain construction. CASTLING uses microarray-synthesized oligonucleotide pools and in vitro recombineering to program the genomic insertion of long DNA constructs via homologous recombination. One simple transformation yields pooled libraries with >90% of correctly tagged clones. Up to several hundred genes can be tagged in a single step and, on a genomic scale, approximately half of all genes are tagged with only ~10-fold oversampling. We report several parameters that affect tagging success and provide a quantitative targeted next-generation sequencing method to analyze such pooled collections. Thus, CASTLING unlocks avenues for increasing throughput in functional genomics and cell biology research.
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
High-throughput functional variant screens via in vivo production of single-stranded DNA.Proc Natl Acad Sci U S A. 2021 May 4;118(18):e2018181118. doi: 10.1073/pnas.2018181118. Proc Natl Acad Sci U S A. 2021. PMID: 33906944 Free PMC article.
-
CRISPR-UnLOCK: Multipurpose Cas9-Based Strategies for Conversion of Yeast Libraries and Strains.Front Microbiol. 2017 Sep 20;8:1773. doi: 10.3389/fmicb.2017.01773. eCollection 2017. Front Microbiol. 2017. PMID: 28979241 Free PMC article.
-
Yeast genetic interaction screens in the age of CRISPR/Cas.Curr Genet. 2019 Apr;65(2):307-327. doi: 10.1007/s00294-018-0887-8. Epub 2018 Sep 25. Curr Genet. 2019. PMID: 30255296 Free PMC article. Review.
-
Enhanced scale and scope of genome engineering and regulation using CRISPR/Cas in Saccharomyces cerevisiae.FEMS Yeast Res. 2019 Nov 1;19(7):foz076. doi: 10.1093/femsyr/foz076. FEMS Yeast Res. 2019. PMID: 31665284 Review.
-
Expanding the CRISPR/Cas9 Toolbox for Gene Engineering in S. cerevisiae.Curr Microbiol. 2020 Mar;77(3):468-478. doi: 10.1007/s00284-019-01851-0. Epub 2020 Jan 4. Curr Microbiol. 2020. PMID: 31901956
Cited by
-
Design and Construction of Portable CRISPR-Cpf1-Mediated Genome Editing in Bacillus subtilis 168 Oriented Toward Multiple Utilities.Front Bioeng Biotechnol. 2020 Sep 2;8:524676. doi: 10.3389/fbioe.2020.524676. eCollection 2020. Front Bioeng Biotechnol. 2020. PMID: 32984297 Free PMC article.
-
Applications of CRISPR/Cas gene-editing technology in yeast and fungi.Arch Microbiol. 2021 Dec 26;204(1):79. doi: 10.1007/s00203-021-02723-7. Arch Microbiol. 2021. PMID: 34954815
-
A colorimetric RT-LAMP assay and LAMP-sequencing for detecting SARS-CoV-2 RNA in clinical samples.Sci Transl Med. 2020 Aug 12;12(556):eabc7075. doi: 10.1126/scitranslmed.abc7075. Epub 2020 Jul 27. Sci Transl Med. 2020. PMID: 32719001 Free PMC article.
-
CRISPR-Cas12a-assisted PCR tagging of mammalian genes.J Cell Biol. 2020 Jun 1;219(6):e201910210. doi: 10.1083/jcb.201910210. J Cell Biol. 2020. PMID: 32406907 Free PMC article.
-
Characterizing Endogenous Protein Complexes with Biological Mass Spectrometry.Chem Rev. 2022 Apr 27;122(8):7386-7414. doi: 10.1021/acs.chemrev.1c00217. Epub 2021 Aug 18. Chem Rev. 2022. PMID: 34406752 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
