

Proteostasis During Cerebral Ischemia
- PMID: 31275110
- PMCID: PMC6594416
- DOI: 10.3389/fnins.2019.00637
Proteostasis During Cerebral Ischemia
Abstract
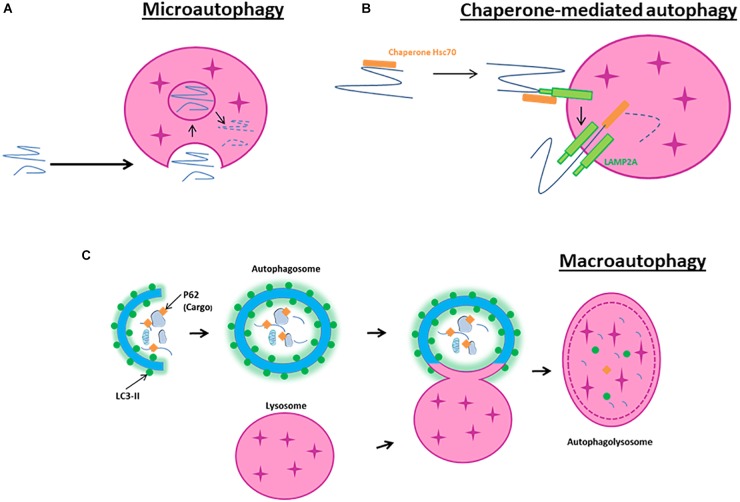
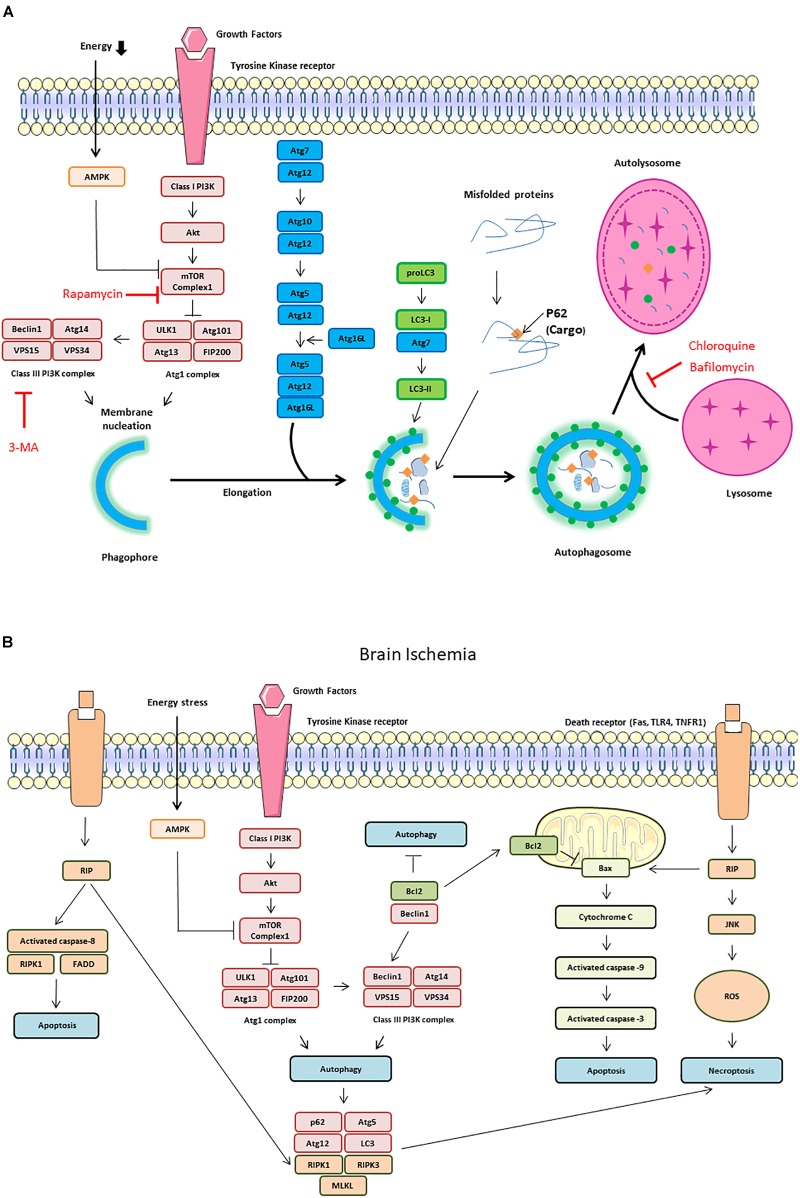
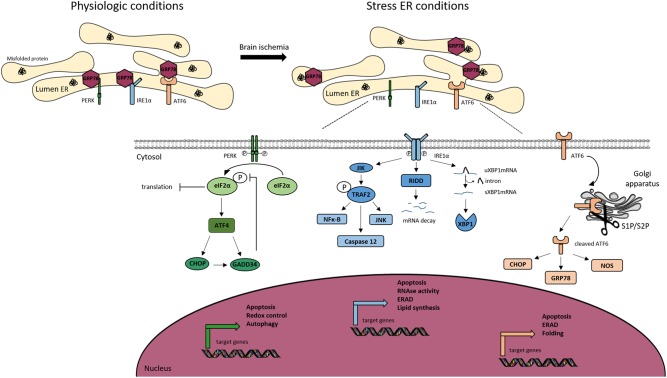
Cerebral ischemia is a complex pathology involving a cascade of cellular mechanisms, which deregulate proteostasis and lead to neuronal death. Proteostasis refers to the equilibrium between protein synthesis, folding, transport, and protein degradation. Within the brain proteostasis plays key roles in learning and memory by controlling protein synthesis and degradation. Two important pathways are implicated in the regulation of proteostasis: the unfolded protein response (UPR) and macroautophagy (called hereafter autophagy). Both are necessary for cell survival, however, their over-activation in duration or intensity can lead to cell death. Moreover, UPR and autophagy can activate and potentiate each other to worsen the issue of cerebral ischemia. A better understanding of autophagy and ER stress will allow the development of therapeutic strategies for stroke, both at the acute phase and during recovery. This review summarizes the latest therapeutic advances implicating ER stress or autophagy in cerebral ischemia. We argue that the processes governing proteostasis should be considered together in stroke, rather than focusing either on ER stress or autophagy in isolation.
Keywords: ER stress; autophagy; mTOR; proteostasis; stroke.
Figures
References
-
- Althausen S., Mengesdorf T., Mies G., Olah L., Nairn A. C., Proud C. G., et al. (2001). Changes in the phosphorylation of initiation factor eIF-2alpha, elongation factor eEF-2 and p70 S6 kinase after transient focal cerebral ischaemia in mice. J. Neurochem. 78 779–787. 10.1046/j.1471-4159.2001.00462.x - DOI - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous