

Disease gene prediction for molecularly uncharacterized diseases
- PMID: 31276496
- PMCID: PMC6636748
- DOI: 10.1371/journal.pcbi.1007078
Disease gene prediction for molecularly uncharacterized diseases
Abstract
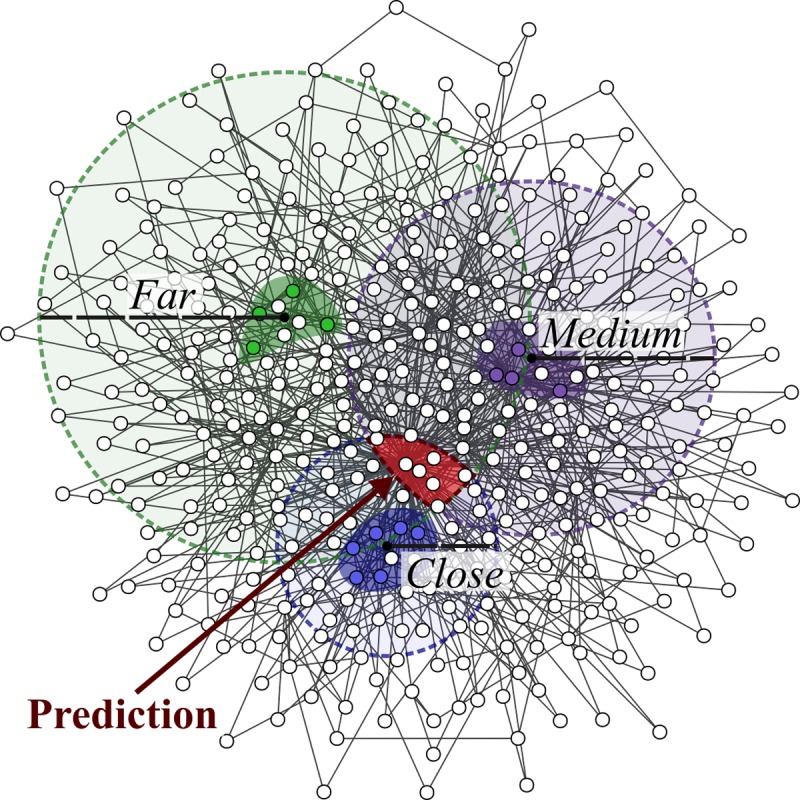
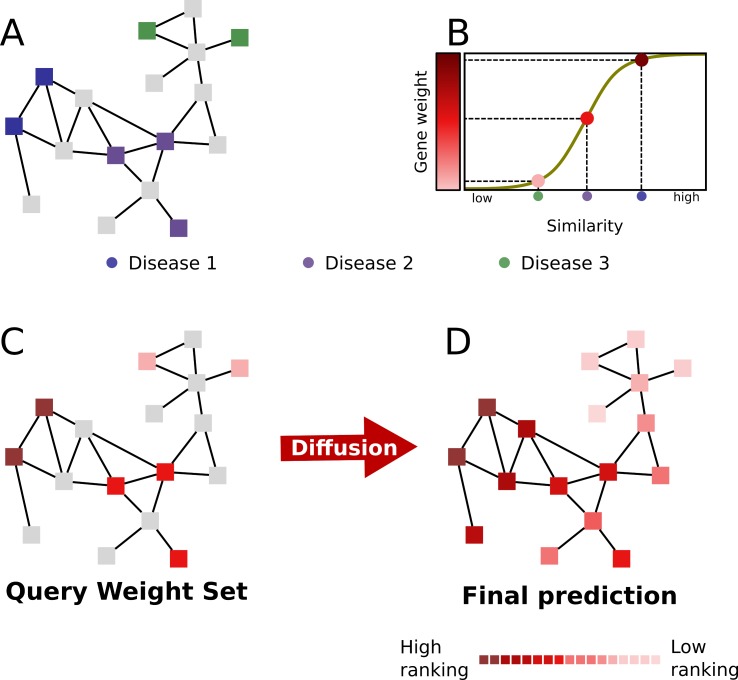
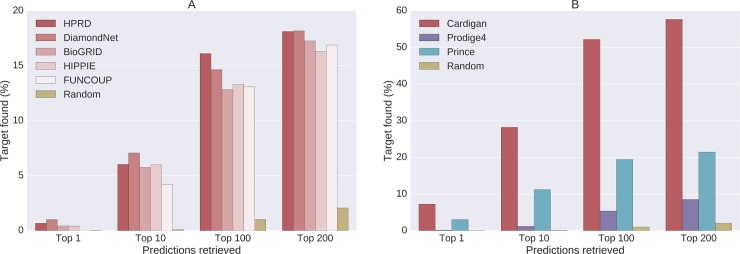
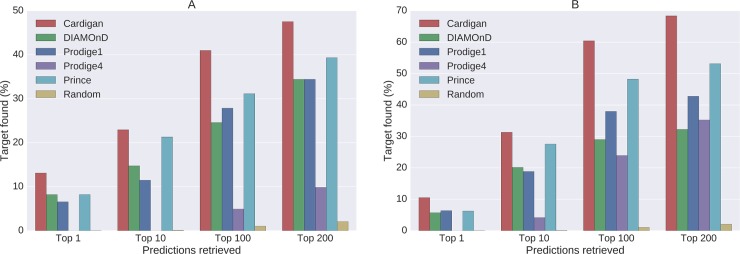
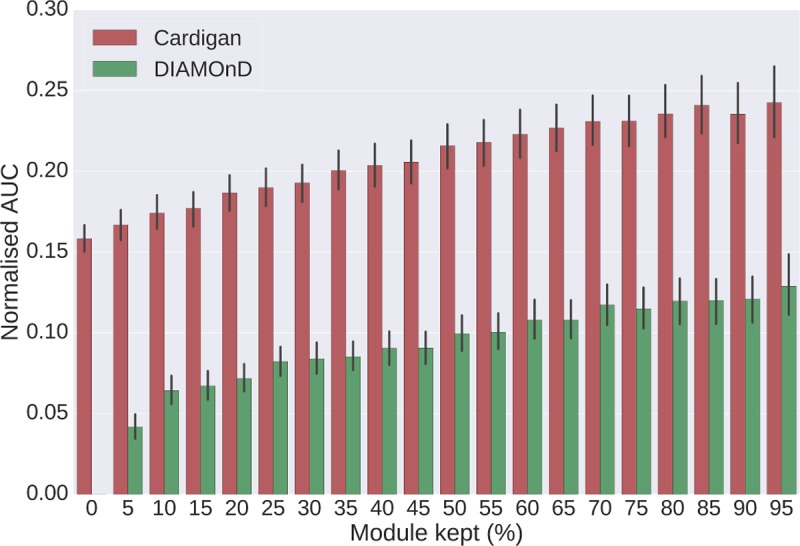
Network medicine approaches have been largely successful at increasing our knowledge of molecularly characterized diseases. Given a set of disease genes associated with a disease, neighbourhood-based methods and random walkers exploit the interactome allowing the prediction of further genes for that disease. In general, however, diseases with no known molecular basis constitute a challenge. Here we present a novel network approach to prioritize gene-disease associations that is able to also predict genes for diseases with no known molecular basis. Our method, which we have called Cardigan (ChARting DIsease Gene AssociatioNs), uses semi-supervised learning and exploits a measure of similarity between disease phenotypes. We evaluated its performance at predicting genes for both molecularly characterized and uncharacterized diseases in OMIM, using both weighted and binary interactomes, and compared it with state-of-the-art methods. Our tests, which use datasets collected at different points in time to replicate the dynamics of the disease gene discovery process, prove that Cardigan is able to accurately predict disease genes for molecularly uncharacterized diseases. Additionally, standard leave-one-out cross validation tests show how our approach outperforms state-of-the-art methods at predicting genes for molecularly characterized diseases by 14%-65%. Cardigan can also be used for disease module prediction, where it outperforms state-of-the-art methods by 87%-299%.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Colhoun HM, McKeigue PM, Smith GD. Problems of reporting genetic associations with complex outcomes. The Lancet. 2003;361(9360):865–72. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
