

De novo substitutions of TRPM3 cause intellectual disability and epilepsy
- PMID: 31278393
- PMCID: PMC6777445
- DOI: 10.1038/s41431-019-0462-x
De novo substitutions of TRPM3 cause intellectual disability and epilepsy
Abstract
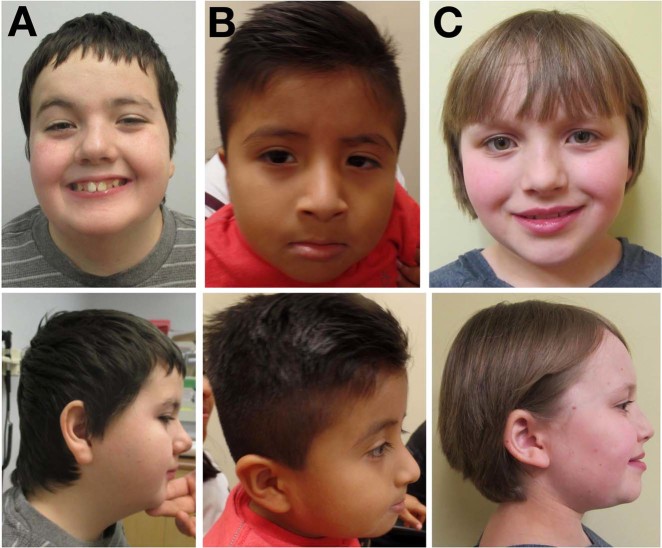
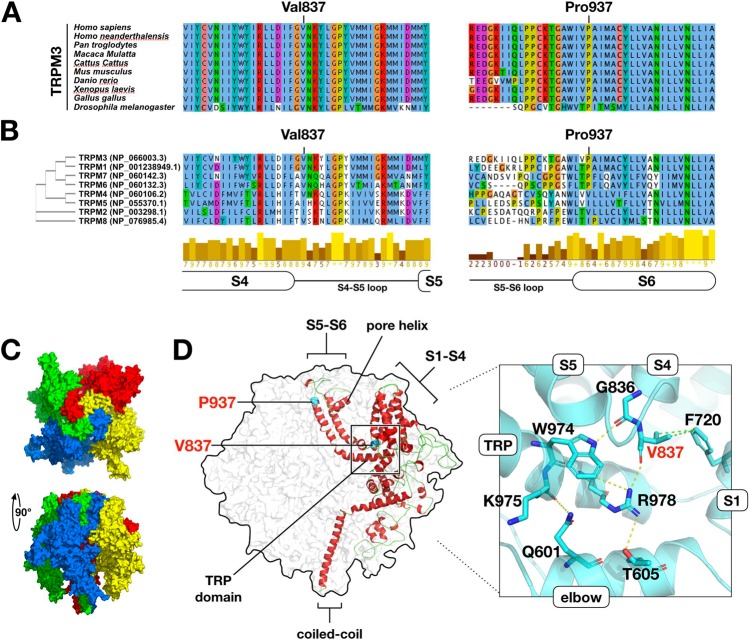
The developmental and epileptic encephalopathies (DEE) are a heterogeneous group of chronic encephalopathies frequently associated with rare de novo nonsynonymous coding variants in neuronally expressed genes. Here, we describe eight probands with a DEE phenotype comprising intellectual disability, epilepsy, and hypotonia. Exome trio analysis showed de novo variants in TRPM3, encoding a brain-expressed transient receptor potential channel, in each. Seven probands were identically heterozygous for a recurrent substitution, p.(Val837Met), in TRPM3's S4-S5 linker region, a conserved domain proposed to undergo conformational change during gated channel opening. The eighth individual was heterozygous for a proline substitution, p.(Pro937Gln), at the boundary between TRPM3's flexible pore-forming loop and an adjacent alpha-helix. General-population truncating variants and microdeletions occur throughout TRPM3, suggesting a pathomechanism other than simple haploinsufficiency. We conclude that de novo variants in TRPM3 are a cause of intellectual disability and epilepsy.
Conflict of interest statement
KMW is an employee of GeneDx, Inc. The other authors declare that they have no conflict of interest.
Figures
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
