

Alternative tumour-specific antigens
- PMID: 31278396
- PMCID: PMC6874891
- DOI: 10.1038/s41568-019-0162-4
Alternative tumour-specific antigens
Abstract
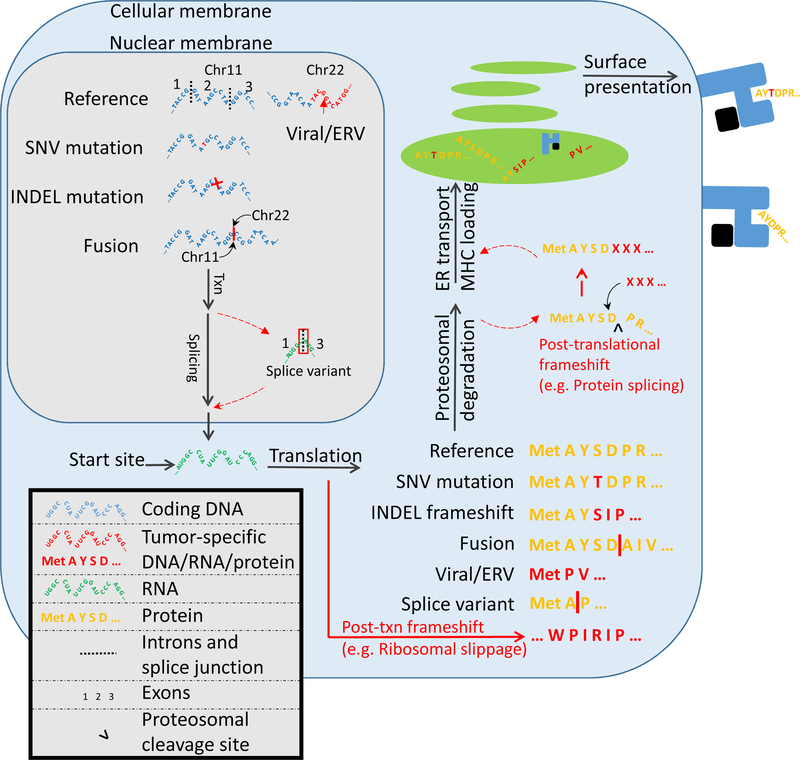
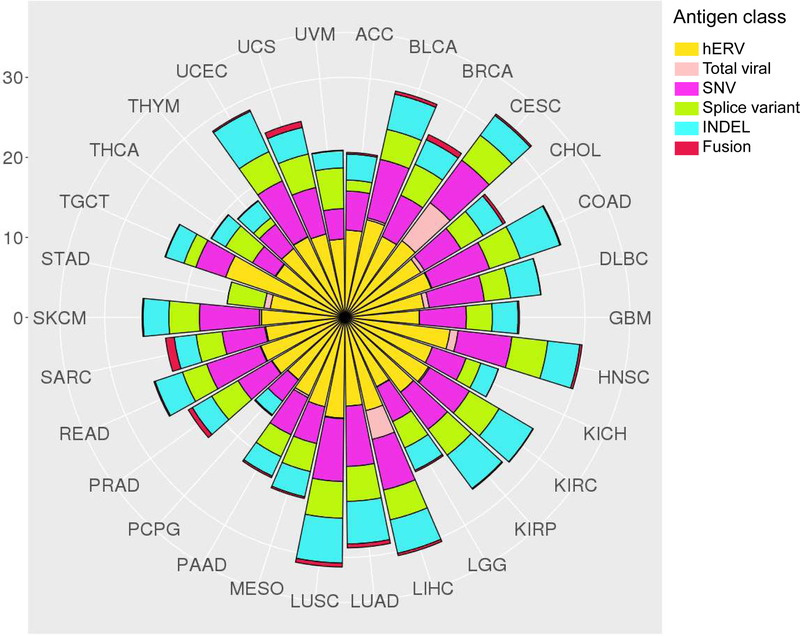
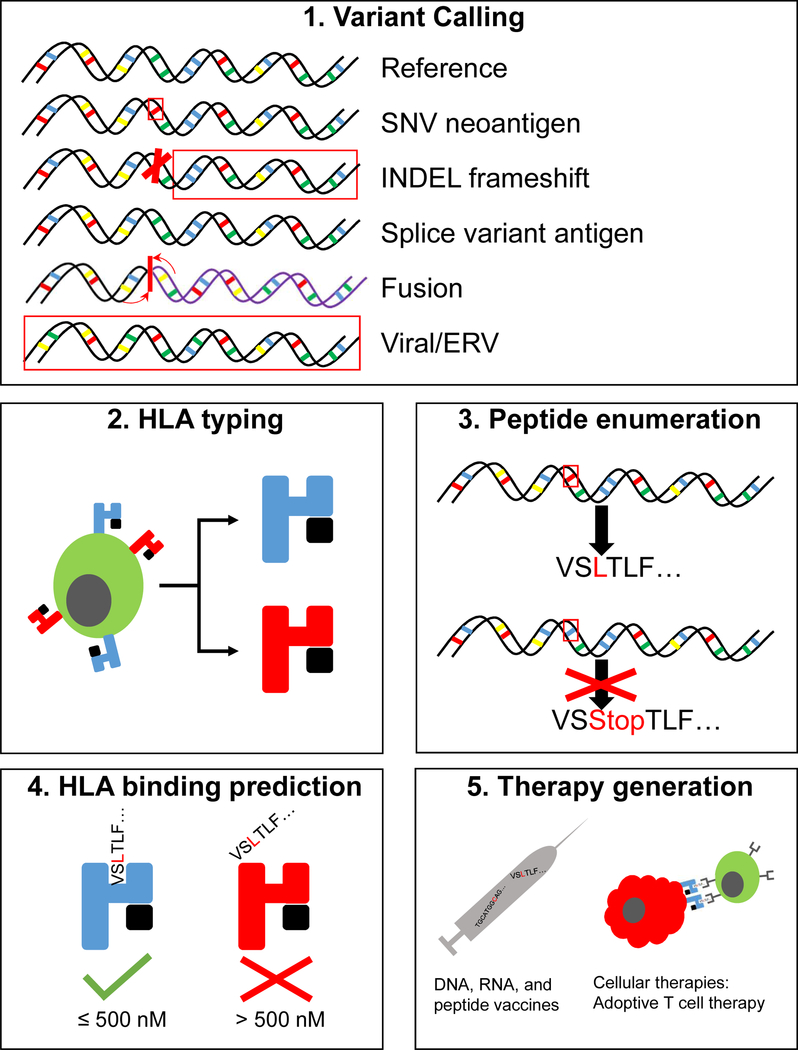
The study of tumour-specific antigens (TSAs) as targets for antitumour therapies has accelerated within the past decade. The most commonly studied class of TSAs are those derived from non-synonymous single-nucleotide variants (SNVs), or SNV neoantigens. However, to increase the repertoire of available therapeutic TSA targets, 'alternative TSAs', defined here as high-specificity tumour antigens arising from non-SNV genomic sources, have recently been evaluated. Among these alternative TSAs are antigens derived from mutational frameshifts, splice variants, gene fusions, endogenous retroelements and other processes. Unlike the patient-specific nature of SNV neoantigens, some alternative TSAs may have the advantage of being widely shared by multiple tumours, allowing for universal, off-the-shelf therapies. In this Opinion article, we will outline the biology, available computational tools, preclinical and/or clinical studies and relevant cancers for each alternative TSA class, as well as discuss both current challenges preventing the therapeutic application of alternative TSAs and potential solutions to aid in their clinical translation.
Figures
References
-
- Sahin U. et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 547, 222–226 (2017). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
