

A heritable subset of the core rumen microbiome dictates dairy cow productivity and emissions
- PMID: 31281883
- PMCID: PMC6609165
- DOI: 10.1126/sciadv.aav8391
A heritable subset of the core rumen microbiome dictates dairy cow productivity and emissions
Abstract
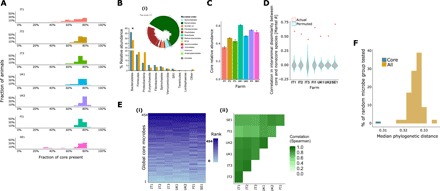
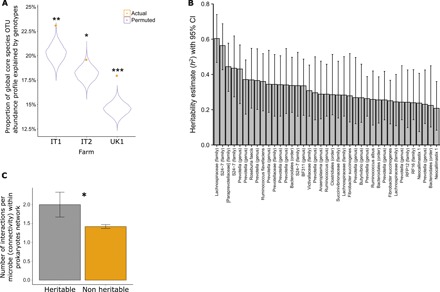
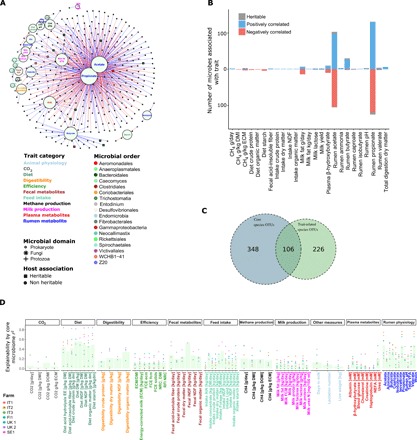
A 1000-cow study across four European countries was undertaken to understand to what extent ruminant microbiomes can be controlled by the host animal and to identify characteristics of the host rumen microbiome axis that determine productivity and methane emissions. A core rumen microbiome, phylogenetically linked and with a preserved hierarchical structure, was identified. A 39-member subset of the core formed hubs in co-occurrence networks linking microbiome structure to host genetics and phenotype (methane emissions, rumen and blood metabolites, and milk production efficiency). These phenotypes can be predicted from the core microbiome using machine learning algorithms. The heritable core microbes, therefore, present primary targets for rumen manipulation toward sustainable and environmentally friendly agriculture.
Figures
References
-
- H. Steinfeld, P. Gerber, T. Wassenaar, V. Caste, M. Rosales, C. de Haan, Livestock’s Long Shadow (FAO, 2006).
-
- A. G. Williams, G. S. Coleman, The Rumen Microbial Ecosystem (Chapman & Hall, 1997).
-
- I. Mizrahi, The Prokaryotes (Springer Berlin Heidelberg, 2013).
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
