

Mutation update: Review of TPP1 gene variants associated with neuronal ceroid lipofuscinosis CLN2 disease
- PMID: 31283065
- PMCID: PMC6851559
- DOI: 10.1002/humu.23860
Mutation update: Review of TPP1 gene variants associated with neuronal ceroid lipofuscinosis CLN2 disease
Abstract
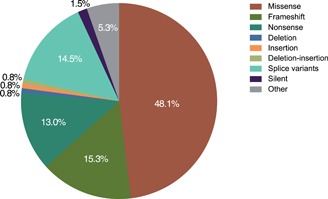
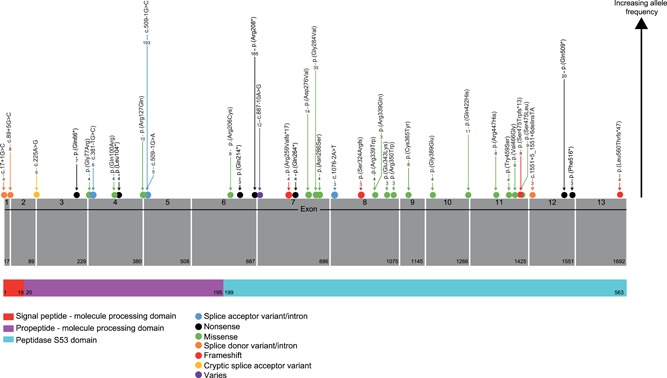
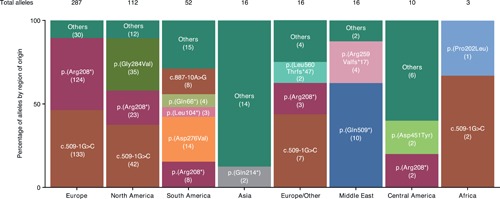
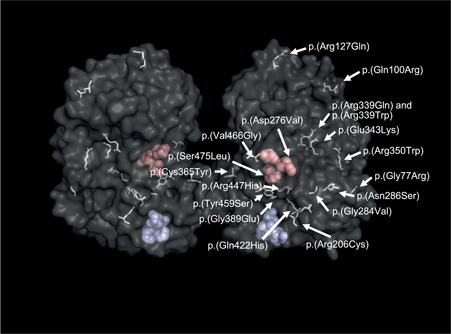
Neuronal ceroid lipofuscinosis type 2 (CLN2 disease) is an autosomal recessive condition caused by variants in the TPP1 gene, leading to deficient activity of the lysosomal enzyme tripeptidyl peptidase I (TPP1). We update on the spectrum of TPP1 variants associated with CLN2 disease, comprising 131 unique variants from 389 individuals (717 alleles) collected from the literature review, public databases, and laboratory communications. Previously unrecorded individuals were added to the UCL TPP1-specific database. Two known pathogenic variants, c.509-1 G>C and c.622 C>T (p.(Arg208*)), collectively occur in 60% of affected individuals in the sample, and account for 50% of disease-associated alleles. At least 86 variants (66%) are private to single families. Homozygosity occurs in 45% of individuals where both alleles are known (87% of reported individuals). Atypical CLN2 disease, TPP1 enzyme deficiency with disease onset and/or progression distinct from classic late-infantile CLN2, represents 13% of individuals recorded with associated phenotype. NCBI ClinVar currently holds records for 37% of variants collected here. Effective CLN2 disease management requires early diagnosis; however, irreversible neurodegeneration occurs before a diagnosis is typically reached at age 5. Timely classification and public reporting of TPP1 variants is essential as molecular testing increases in use as a first-line diagnostic test for pediatric-onset neurological disease.
Keywords: genotype-phenotype correlation; late-infantile neuronal ceroid lipofuscinosis; lysosomal storage disorders; neurodegeneration; tripeptidyl peptidase I.
© 2019 The Authors. Human Mutation Published by Wiley Periodicals, Inc.
Conflict of interest statement
A. Schulz has received personal fees from BioMarin Pharmaceutical Inc., outside of the submitted work. M. Aristorena has no conflicts of interest to declare. M. Bailey was an employee of BioMarin Pharmaceutical Inc. at the time of the study. N. Miller was an employee of BioMarin Pharmaceutical Inc. at the time of the study. Professor S. E. Mole receives financial support from BioMarin Pharmaceutical Inc. to maintain the NCL Mutation Database and acts as an advisor to BioMarin Pharmaceutical Inc. on mutations in
Figures
References
-
- Awano, T. , Katz, M. L. , O'brien, D. P. , Sohar, I. , Lobel, P. , Coates, J. R. , … Johnson, G. S. (2006). A frame shift mutation in canine TPP1 (the ortholog of human CLN2) in a juvenile Dachshund with neuronal ceroid lipofuscinosis. Molecular Genetics and Metabolism, 89(3), 254–260. 10.1016/j.ymgme.2006.02.016 - DOI - PubMed
-
- Barcenas, M. , Xue, C. , Marushchak‐Vlaskin, T. , Scott, C. R. , Gelb, M. H. , & Tureček, F. (2014). Tandem mass spectrometry assays of palmitoyl protein thioesterase 1 and tripeptidyl peptidase activity in dried blood spots for the detection of neuronal ceroid lipofuscinoses in newborns. Analytical Chemistry, 86(15), 7962–7968. 10.1021/ac501994b - DOI - PMC - PubMed
-
- Van Beersel, G. , Tihon, E. , Demine, S. , Hamer, I. , Jadot, M. , & Arnould, T. (2013). Different molecular mechanisms involved in spontaneous and oxidative stress‐induced mitochondrial fragmentation in tripeptidyl peptidase‐1 (TPP‐1)‐deficient fibroblasts. Bioscience Reports, 33(2), e00023 10.1042/BSR20120104 - DOI - PMC - PubMed
-
- Berry‐Kravis, E. , Sleat, D. E. , Sohar, I. , Meyer, P. , Donnelly, R. , & Lobel, P. (2000). Prenatal testing for late infantile neuronal ceroid lipofuscinosis. Annals of Neurology, 47(2), 254–257. - PubMed
-
- Bhavsar, R. , Mistri, M. , Kamate, M. , Shah, R. , Mehta, S. , Shah, H. , & Sheth, J. (2016). Clinical presentation and molecular characterization of children with neuronal ceroid lipofuscinosis (NCL I & II) from India. Journal of Inherited Metabolic Disease, 39(Suppl 1), S35–S284. 10.1007/s10545-016-9969-2 - DOI
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
