

Monitoring Cell-Type-Specific Gene Expression Using Ribosome Profiling In Vivo During Cardiac Hemodynamic Stress
- PMID: 31284834
- PMCID: PMC6690133
- DOI: 10.1161/CIRCRESAHA.119.314817
Monitoring Cell-Type-Specific Gene Expression Using Ribosome Profiling In Vivo During Cardiac Hemodynamic Stress
Abstract
Rationale: Gene expression profiles have been mainly determined by analysis of transcript abundance. However, these analyses cannot capture posttranscriptional gene expression control at the level of translation, which is a key step in the regulation of gene expression, as evidenced by the fact that transcript levels often poorly correlate with protein levels. Furthermore, genome-wide transcript profiling of distinct cell types is challenging due to the fact that lysates from tissues always represent a mixture of cells.
Objectives: This study aimed to develop a new experimental method that overcomes both limitations and to apply this method to perform a genome-wide analysis of gene expression on the translational level in response to pressure overload.
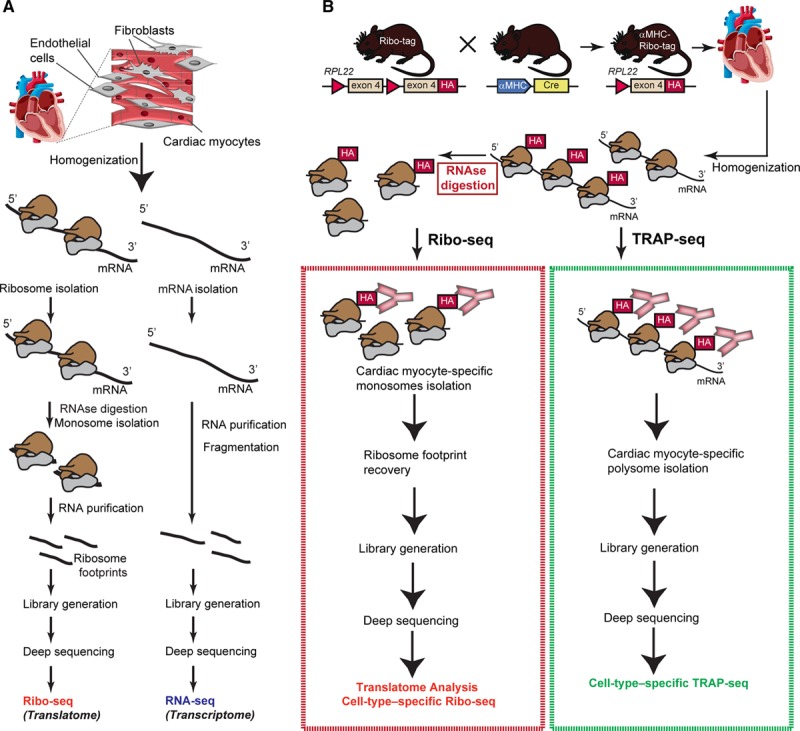
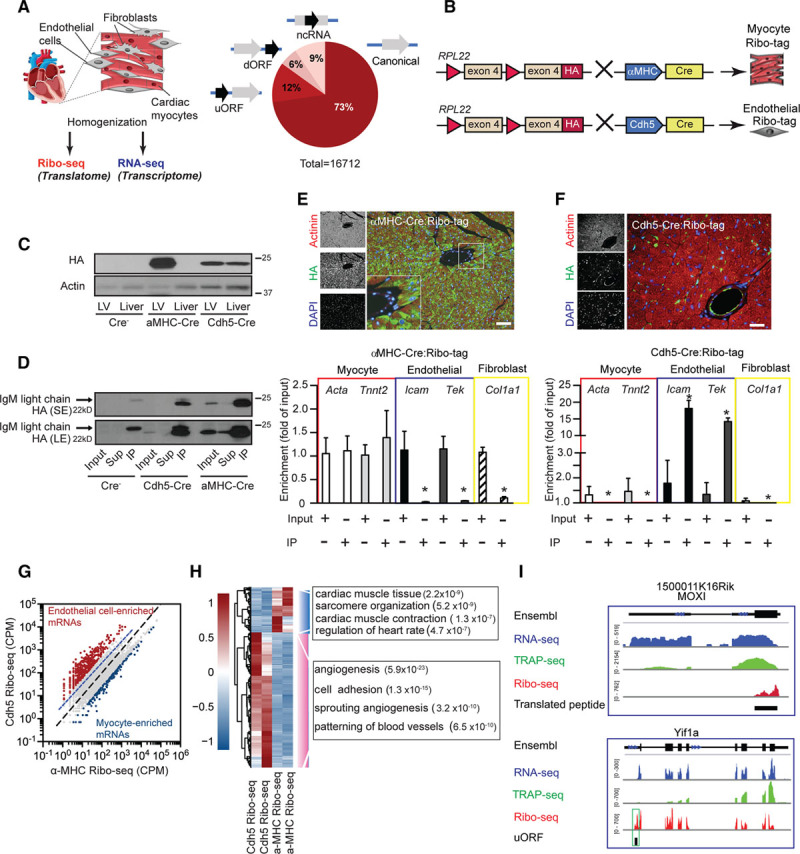
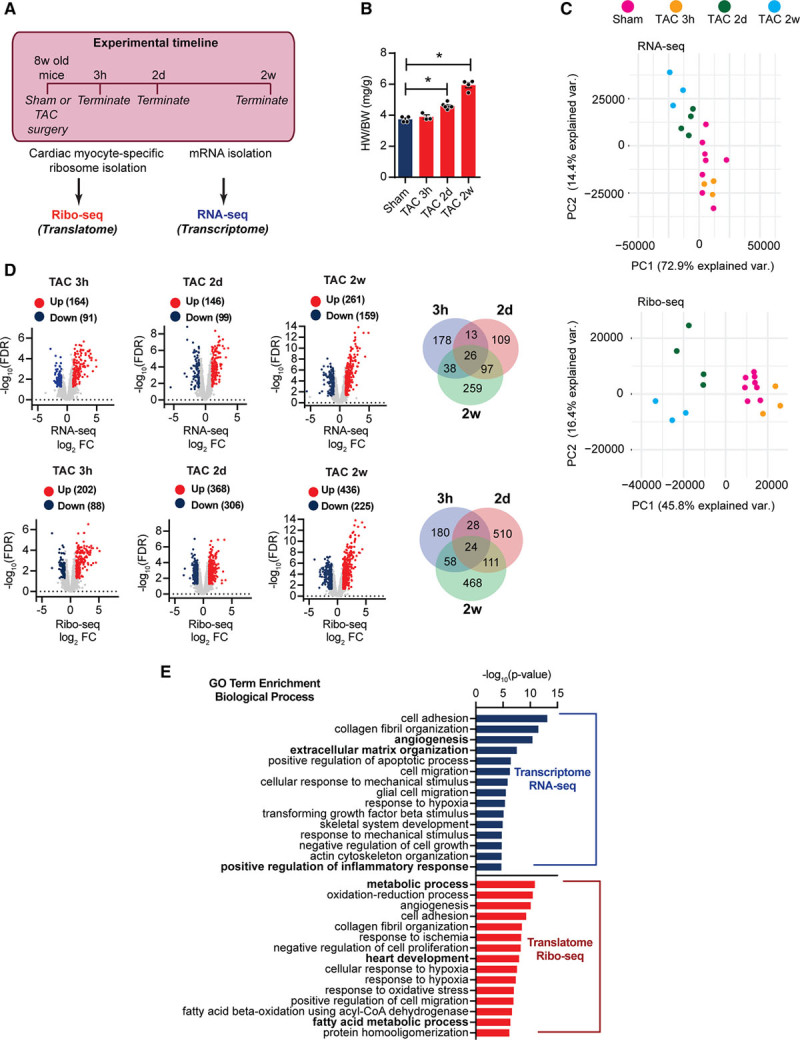
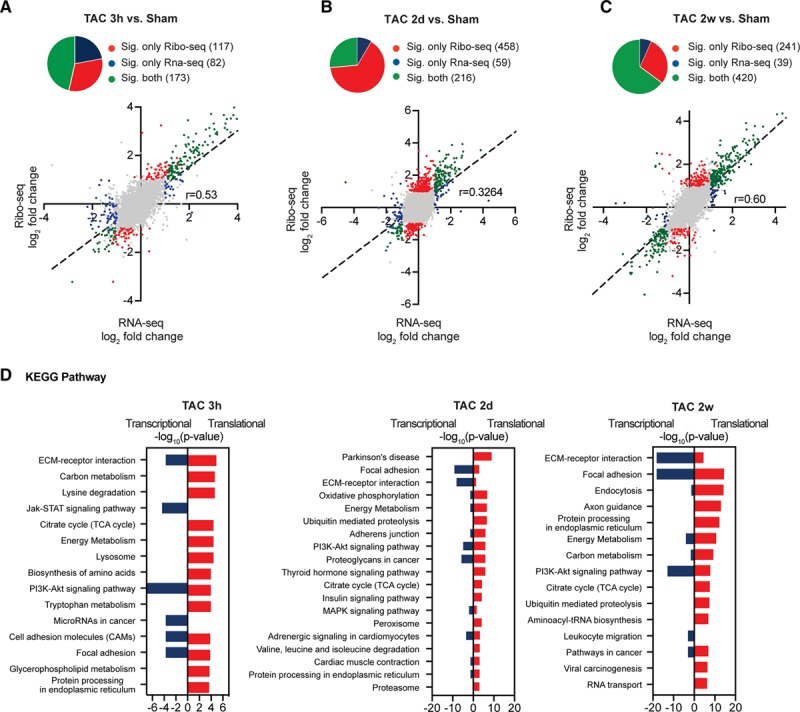
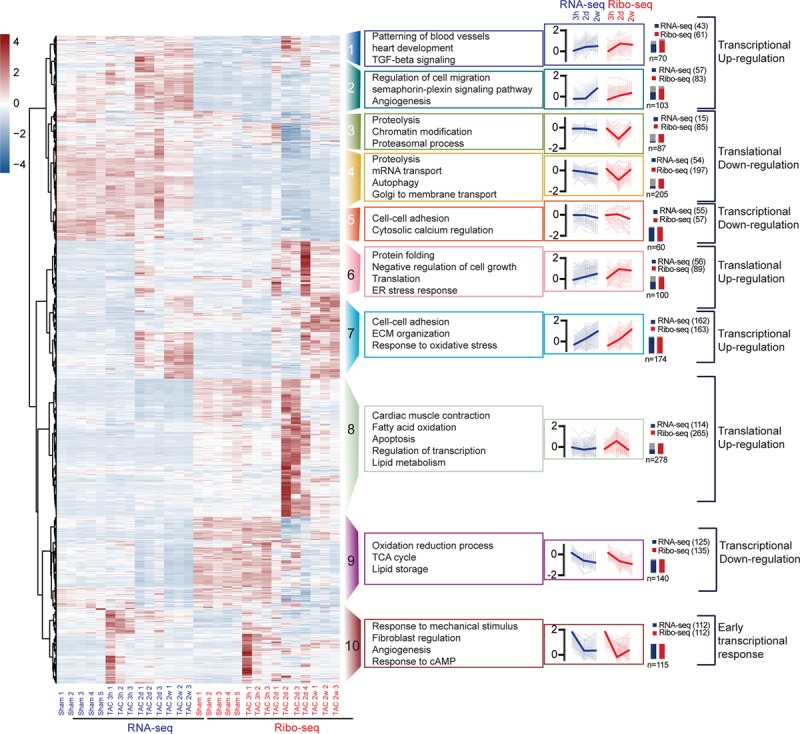
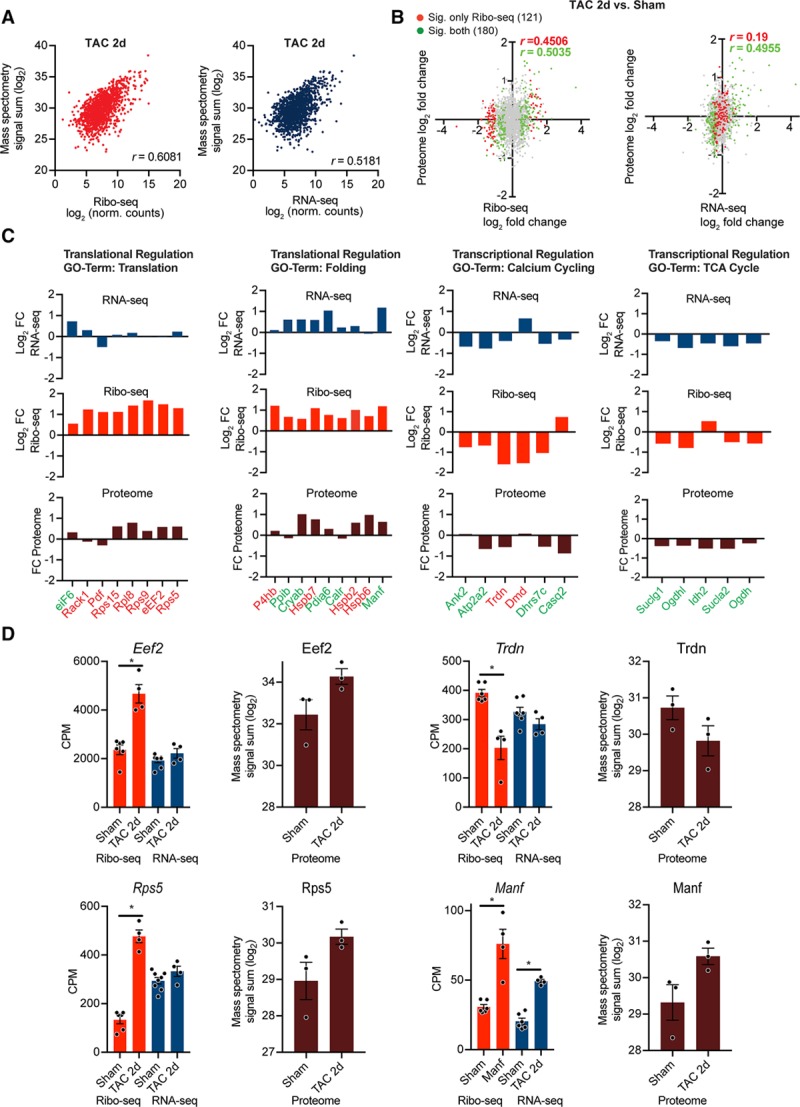
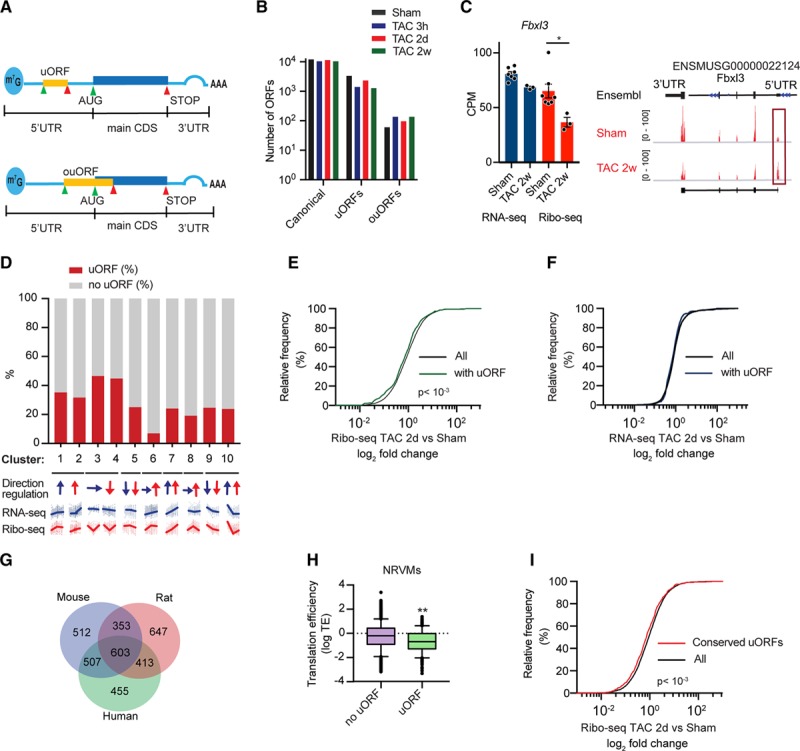
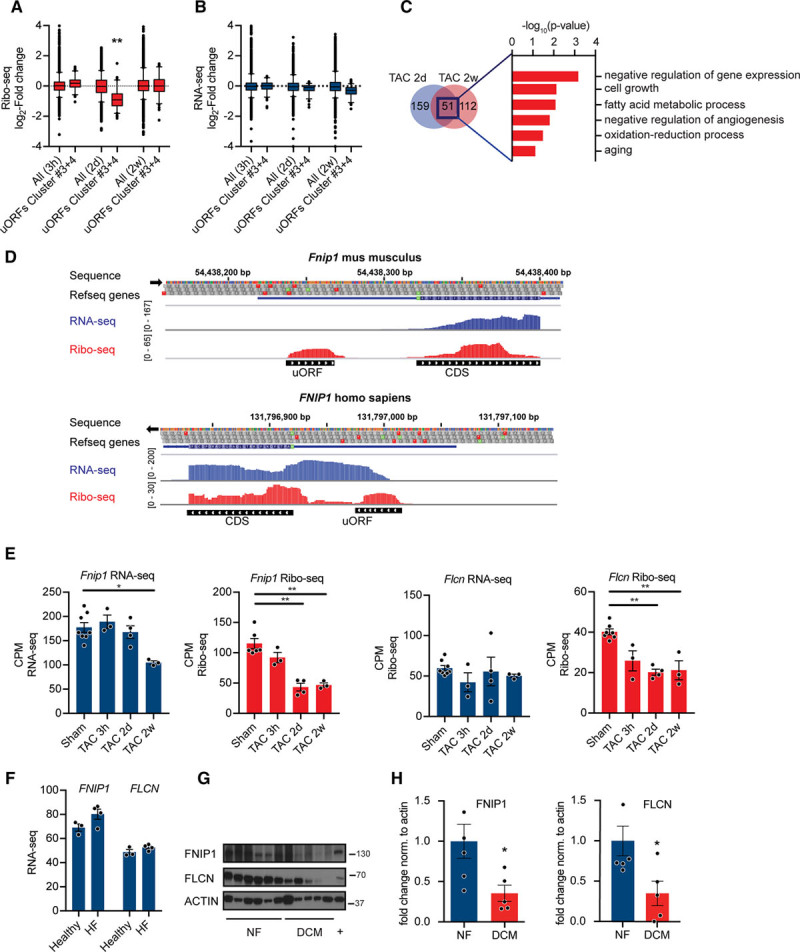
Methods and results: By combining ribosome profiling (Ribo-seq) with a ribosome-tagging approach (Ribo-tag), it was possible to determine the translated transcriptome in specific cell types from the heart. After pressure overload, we monitored the cardiac myocyte translatome by purifying tagged cardiac myocyte ribosomes from cardiac lysates and subjecting the ribosome-protected mRNA fragments to deep sequencing. We identified subsets of mRNAs that are regulated at the translational level and found that translational control determines early changes in gene expression in response to cardiac stress in cardiac myocytes. Translationally controlled transcripts are associated with specific biological processes related to translation, protein quality control, and metabolism. Mechanistically, Ribo-seq allowed for the identification of upstream open reading frames in transcripts, which we predict to be important regulators of translation.
Conclusions: This method has the potential to (1) provide a new tool for studying cell-specific gene expression at the level of translation in tissues, (2) reveal new therapeutic targets to prevent cellular remodeling, and (3) trigger follow-up studies that address both, the molecular mechanisms involved in the posttranscriptional control of gene expression in cardiac cells, and the protective functions of proteins expressed in response to cellular stress.
Keywords: gene expression; hypertrophy, left ventricular; metabolism; protein biosynthesis; protein folding; proteostasis; ribosomes.
Figures
References
-
- Mudd JO, Kass DA. Tackling heart failure in the twenty-first century. Nature. 2008;451:919–928. doi: 10.1038/nature06798. - PubMed
-
- Preissl S, Schwaderer M, Raulf A, Hesse M, Grüning BA, Köbele C, Backofen R, Fleischmann BK, Hein L, Gilsbach R. Deciphering the epigenetic code of cardiac myocyte transcription. Circ Res. 2015;117:413–423. doi: 10.1161/CIRCRESAHA.115.306337. - PubMed
-
- Thum T, Condorelli G. Long noncoding RNAs and microRNAs in cardiovascular pathophysiology. Circ Res. 2015;116:751–762. doi: 10.1161/CIRCRESAHA.116.303549. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
