

In vivo stabilization of OPA1 in hepatocytes potentiates mitochondrial respiration and gluconeogenesis in a prohibitin-dependent way
- PMID: 31285263
- PMCID: PMC6709633
- DOI: 10.1074/jbc.RA119.007601
In vivo stabilization of OPA1 in hepatocytes potentiates mitochondrial respiration and gluconeogenesis in a prohibitin-dependent way
Abstract
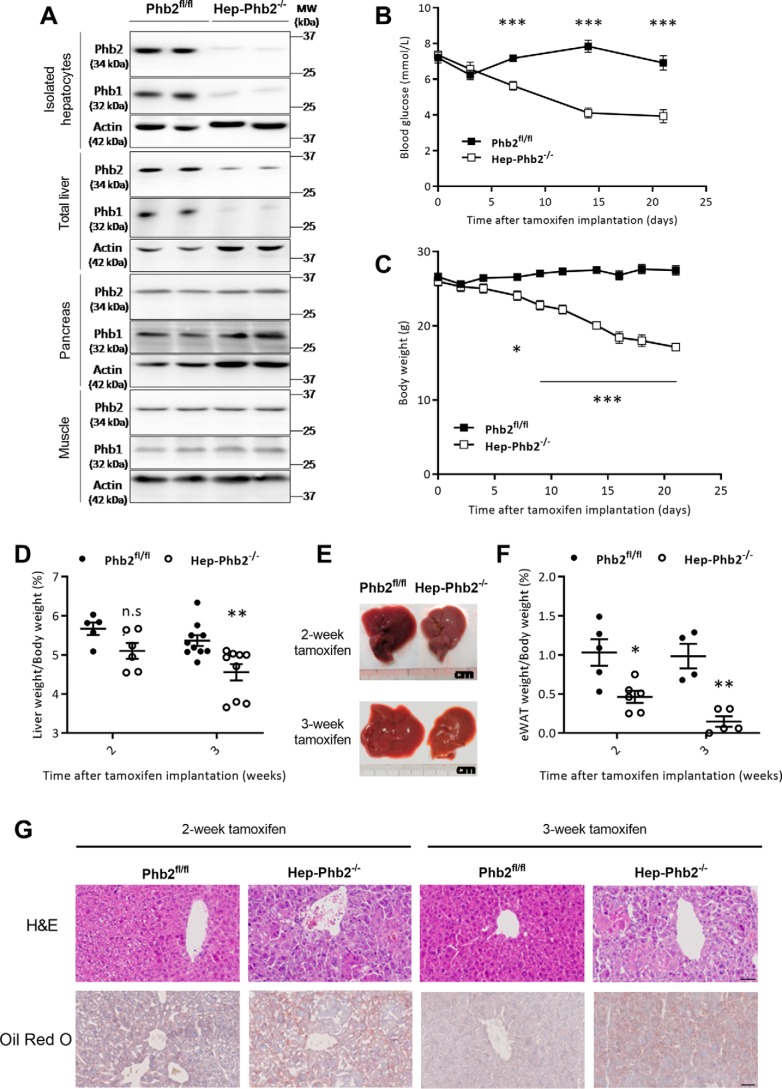
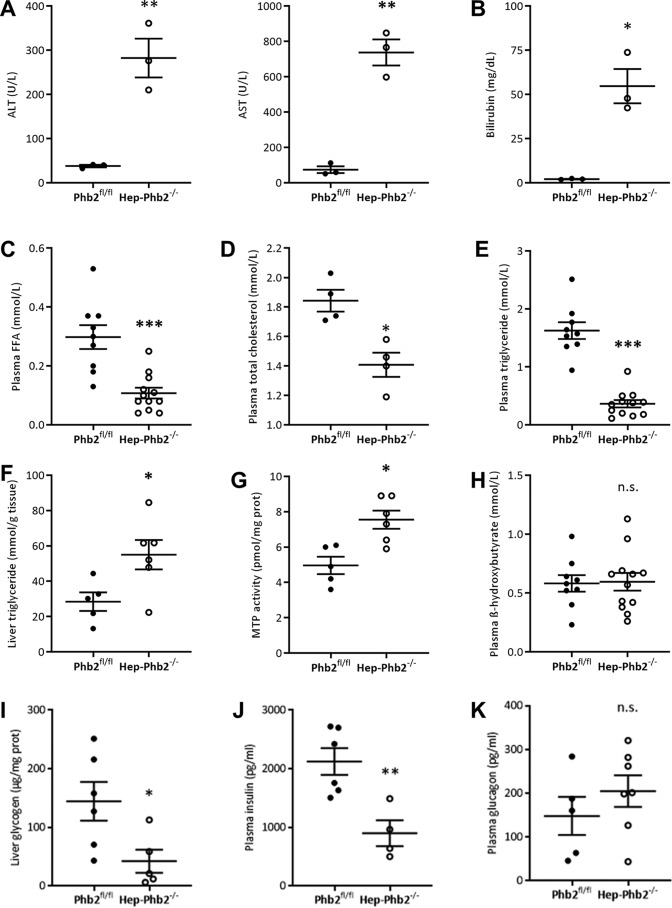
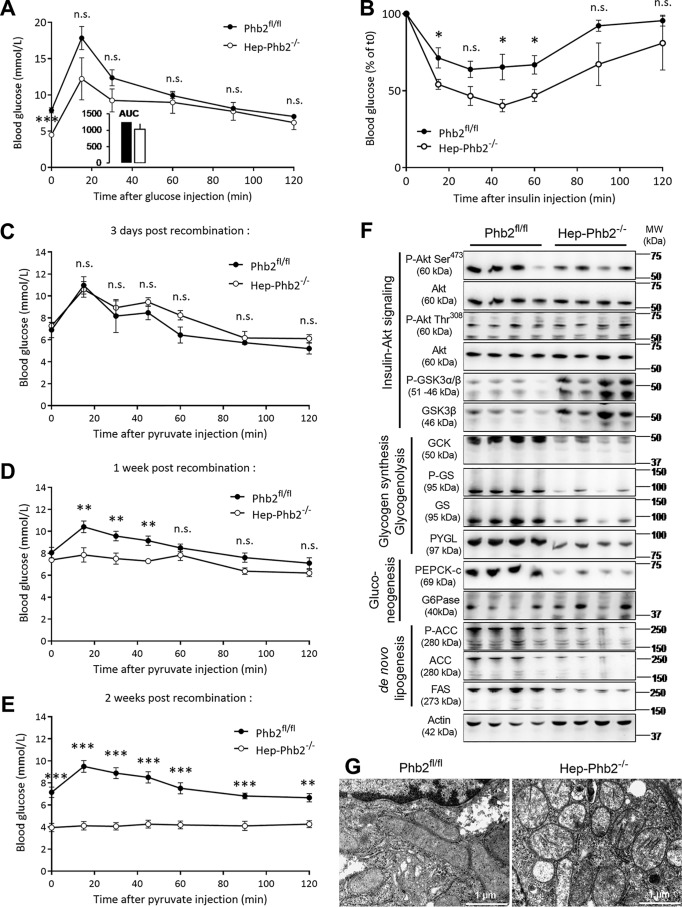
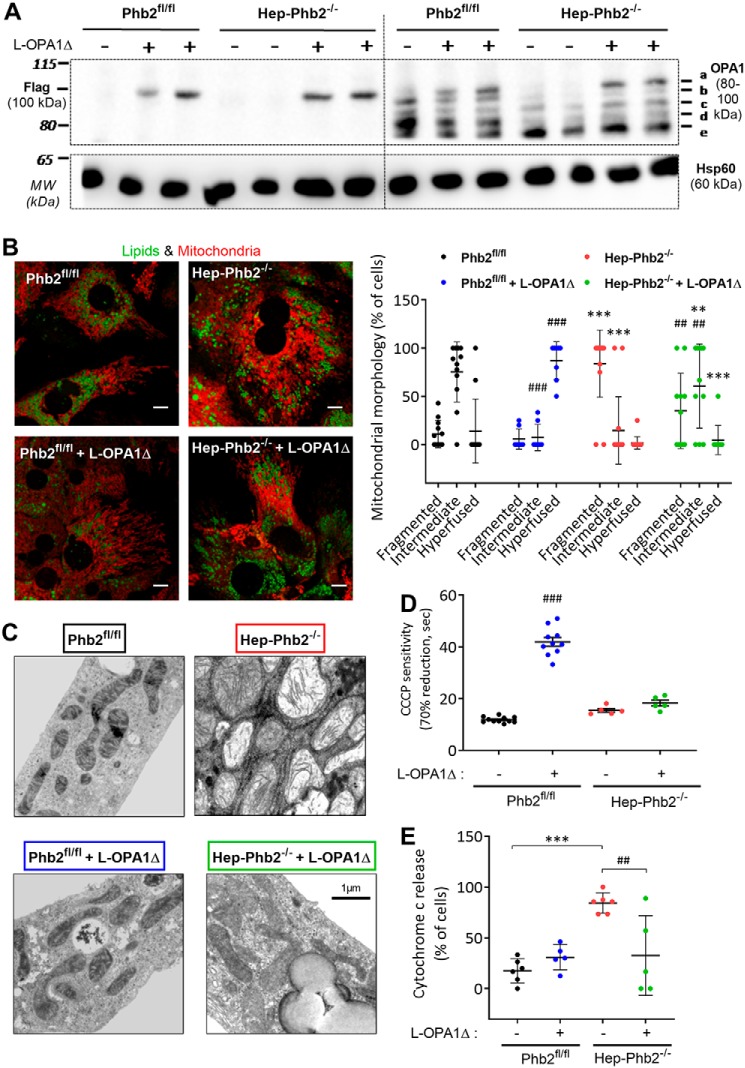
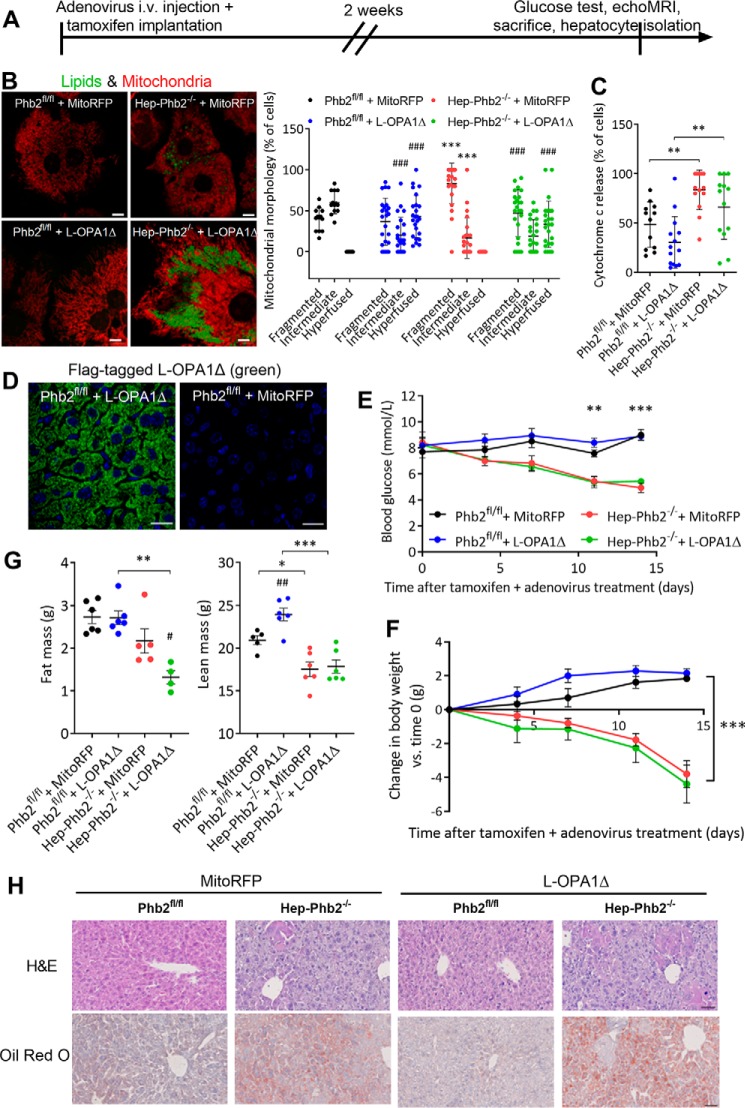
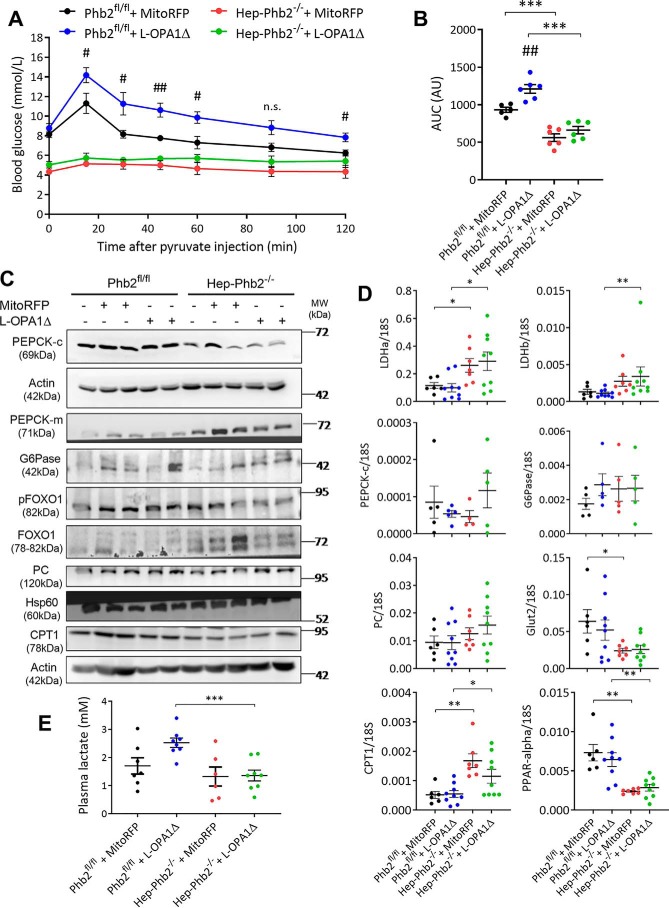
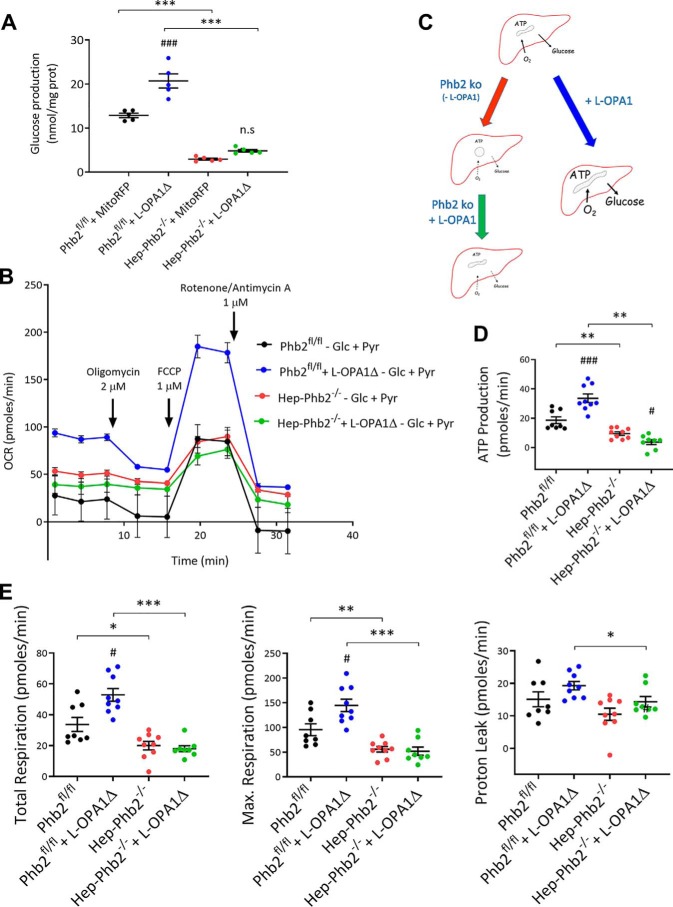
Patients with fatty liver diseases present altered mitochondrial morphology and impaired metabolic function. Mitochondrial dynamics and related cell function require the uncleaved form of the dynamin-like GTPase OPA1. Stabilization of OPA1 might then confer a protective mechanism against stress-induced tissue damages. To study the putative role of hepatic mitochondrial morphology in a sick liver, we expressed a cleavage-resistant long form of OPA1 (L-OPA1Δ) in the liver of a mouse model with mitochondrial liver dysfunction (i.e. the hepatocyte-specific prohibitin-2 knockout (Hep-Phb2-/-) mice). Liver prohibitin-2 deficiency caused excessive proteolytic cleavage of L-OPA1, mitochondrial fragmentation, and increased apoptosis. These molecular alterations were associated with lipid accumulation, abolished gluconeogenesis, and extensive liver damage. Such liver dysfunction was associated with severe hypoglycemia. In prohibitin-2 knockout mice, expression of L-OPA1Δ by in vivo adenovirus delivery restored the morphology but not the function of mitochondria in hepatocytes. In prohibitin-competent mice, elongation of liver mitochondria by expression of L-OPA1Δ resulted in excessive glucose production associated with increased mitochondrial respiration. In conclusion, mitochondrial dynamics participates in the control of hepatic glucose production.
Keywords: OPA1; gluconeogenesis; hepatocyte; liver; liver metabolism; mitochondria; mitochondrial metabolism; prohibitins.
© 2019 Li et al.
Conflict of interest statement
The authors declare that they have no conflicts of interest with the contents of this article
Figures
Comment in
-
Phb1:Phb2 heterodimers in the mitochondria-beyond functional interdependence.J Biol Chem. 2019 Oct 4;294(40):14836. doi: 10.1074/jbc.L119.010788. J Biol Chem. 2019. PMID: 31586026 Free PMC article. No abstract available.
-
Reply to Mishra: Prohibitin heterodimers-a complex time dependence for carcinogenesis.J Biol Chem. 2019 Oct 4;294(40):14837. doi: 10.1074/jbc.RL119.010819. J Biol Chem. 2019. PMID: 31586027 Free PMC article. No abstract available.
Similar articles
-
Loss of OMA1 delays neurodegeneration by preventing stress-induced OPA1 processing in mitochondria.J Cell Biol. 2016 Jan 18;212(2):157-66. doi: 10.1083/jcb.201507022. J Cell Biol. 2016. PMID: 26783299 Free PMC article.
-
Bif-1 Interacts with Prohibitin-2 to Regulate Mitochondrial Inner Membrane during Cell Stress and Apoptosis.J Am Soc Nephrol. 2019 Jul;30(7):1174-1191. doi: 10.1681/ASN.2018111117. Epub 2019 May 24. J Am Soc Nephrol. 2019. PMID: 31126972 Free PMC article.
-
Prohibitins control cell proliferation and apoptosis by regulating OPA1-dependent cristae morphogenesis in mitochondria.Genes Dev. 2008 Feb 15;22(4):476-88. doi: 10.1101/gad.460708. Genes Dev. 2008. PMID: 18281461 Free PMC article.
-
Prohibitins and the functional compartmentalization of mitochondrial membranes.J Cell Sci. 2009 Nov 1;122(Pt 21):3823-30. doi: 10.1242/jcs.037655. J Cell Sci. 2009. PMID: 19889967 Review.
-
Targeted OMA1 therapies for cancer.Int J Cancer. 2019 Nov 1;145(9):2330-2341. doi: 10.1002/ijc.32177. Epub 2019 Feb 21. Int J Cancer. 2019. PMID: 30714136 Review.
Cited by
-
PHB2 promotes SHIP2 ubiquitination via the E3 ligase NEDD4 to regulate AKT signaling in gastric cancer.J Exp Clin Cancer Res. 2024 Jan 11;43(1):17. doi: 10.1186/s13046-023-02937-1. J Exp Clin Cancer Res. 2024. PMID: 38200519 Free PMC article.
-
Phb1:Phb2 heterodimers in the mitochondria-beyond functional interdependence.J Biol Chem. 2019 Oct 4;294(40):14836. doi: 10.1074/jbc.L119.010788. J Biol Chem. 2019. PMID: 31586026 Free PMC article. No abstract available.
-
Prohibitin 2 deficiency impairs cardiac fatty acid oxidation and causes heart failure.Cell Death Dis. 2020 Mar 12;11(3):181. doi: 10.1038/s41419-020-2374-7. Cell Death Dis. 2020. PMID: 32165613 Free PMC article.
-
Role of mitochondrial quality control in the pathogenesis of nonalcoholic fatty liver disease.Aging (Albany NY). 2020 Mar 26;12(7):6467-6485. doi: 10.18632/aging.102972. Epub 2020 Mar 26. Aging (Albany NY). 2020. PMID: 32213662 Free PMC article. Review.
-
Genome-wide CRISPRi screening identifies OCIAD1 as a prohibitin client and regulatory determinant of mitochondrial Complex III assembly in human cells.Elife. 2021 May 26;10:e67624. doi: 10.7554/eLife.67624. Elife. 2021. PMID: 34034859 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
