

Potential of nintedanib in treatment of progressive fibrosing interstitial lung diseases
- PMID: 31285305
- PMCID: PMC6751387
- DOI: 10.1183/13993003.00161-2019
Potential of nintedanib in treatment of progressive fibrosing interstitial lung diseases
Abstract
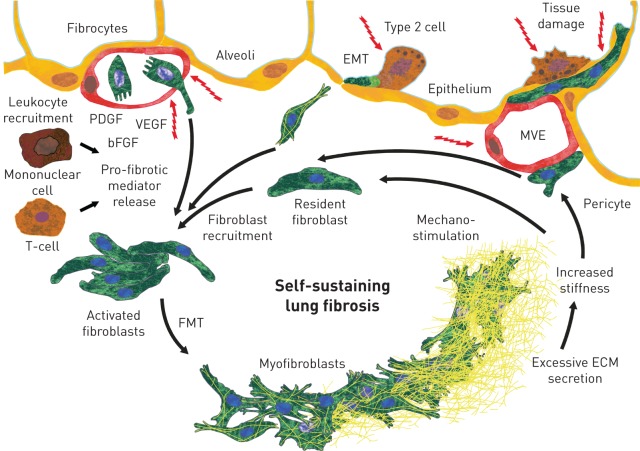
A proportion of patients with fibrosing interstitial lung diseases (ILDs) develop a progressive phenotype characterised by decline in lung function, worsening quality of life and early mortality. Other than idiopathic pulmonary fibrosis (IPF), there are no approved drugs for fibrosing ILDs and a poor evidence base to support current treatments. Fibrosing ILDs with a progressive phenotype show commonalities in clinical behaviour and in the pathogenic mechanisms that drive disease worsening. Nintedanib is an intracellular inhibitor of tyrosine kinases that has been approved for treatment of IPF and has recently been shown to reduce the rate of lung function decline in patients with ILD associated with systemic sclerosis (SSc-ILD). In vitro data demonstrate that nintedanib inhibits several steps in the initiation and progression of lung fibrosis, including the release of pro-inflammatory and pro-fibrotic mediators, migration and differentiation of fibrocytes and fibroblasts, and deposition of extracellular matrix. Nintedanib also inhibits the proliferation of vascular cells. Studies in animal models with features of fibrosing ILDs such as IPF, SSc-ILD, rheumatoid arthritis-ILD, hypersensitivity pneumonitis and silicosis demonstrate that nintedanib has anti-fibrotic activity irrespective of the trigger for the lung pathology. This suggests that nintedanib inhibits fundamental processes in the pathogenesis of fibrosis. A trial of nintedanib in patients with progressive fibrosing ILDs other than IPF (INBUILD) will report results in 2019.
Copyright ©ERS 2019.
Conflict of interest statement
Conflict of interest: L. Wollin is an employee of Boehringer Ingelheim Pharma GmbH & Co. KG. Conflict of interest: J.H.W. Distler has nothing to disclose. Conflict of interest: E.F. Redente has nothing to disclose. Conflict of interest: D.W.H. Riches has nothing to disclose. Conflict of interest: S. Stowasser is an employee of Boehringer Ingelheim International GmbH. Conflict of interest: R. Schlenker-Herceg is an employee of Boehringer Ingelheim Pharmaceuticals, Inc. Conflict of interest: T.M. Maher has, via his institution, received industry-academic funding from GlaxoSmithKline R&D and UCB; has received consultancy or speakers fees from Apellis, AstraZeneca, Bayer, Biogen Idec, Boehringer Ingelheim, Galapagos, GlaxoSmithKline R&D, Indalo, Pliant, ProMetic, Roche, Samumed and UCB; and has received consultancy fees from Galecto. Conflict of interest: M. Kolb reports grants and personal fees for consultancy and lecturing from Roche and Boehringer Ingelheim, grants and personal fees for consultancy from GSK, Gilead and Prometic, grants from Actelion, Respivert, Alkermes and Pharmaxis, personal fees for consultancy from Genoa, Indalo and Third Pole, outside the submitted work.
Figures
References
-
- Raghu G, Remy-Jardin M, Myers JL, et al. . Diagnosis of idiopathic pulmonary fibrosis. An official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med 2018; 198: e44–e68. - PubMed
-
- Wells AU, Brown KK, Flaherty KR, et al. . What's in a name? That which we call IPF, by any other name would act the same. Eur Respir J 2018; 51: 1800692. - PubMed
-
- Jegal Y, Kim DS, Shim TS, et al. . Physiology is a stronger predictor of survival than pathology in fibrotic interstitial pneumonia. Am J Respir Crit Care Med 2005; 171: 639–644. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical