

Chemical genomics reveals histone deacetylases are required for core regulatory transcription
- PMID: 31285436
- PMCID: PMC6614369
- DOI: 10.1038/s41467-019-11046-7
Chemical genomics reveals histone deacetylases are required for core regulatory transcription
Abstract
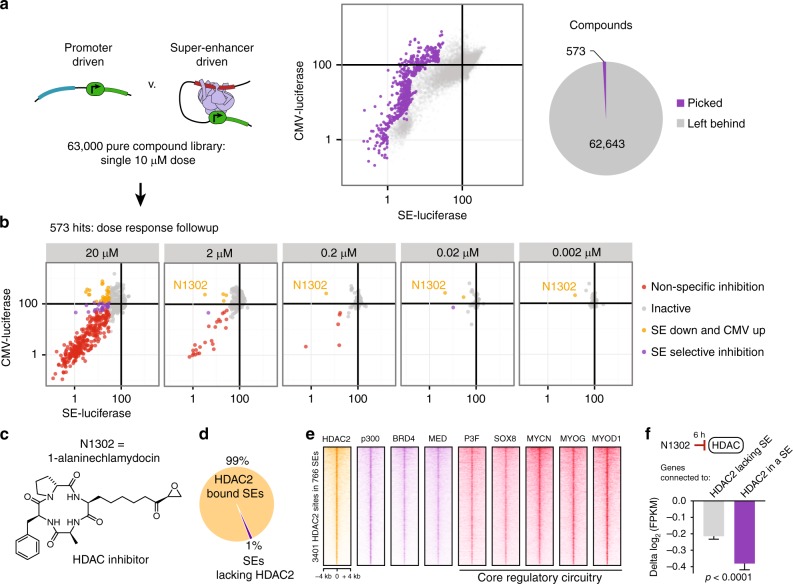
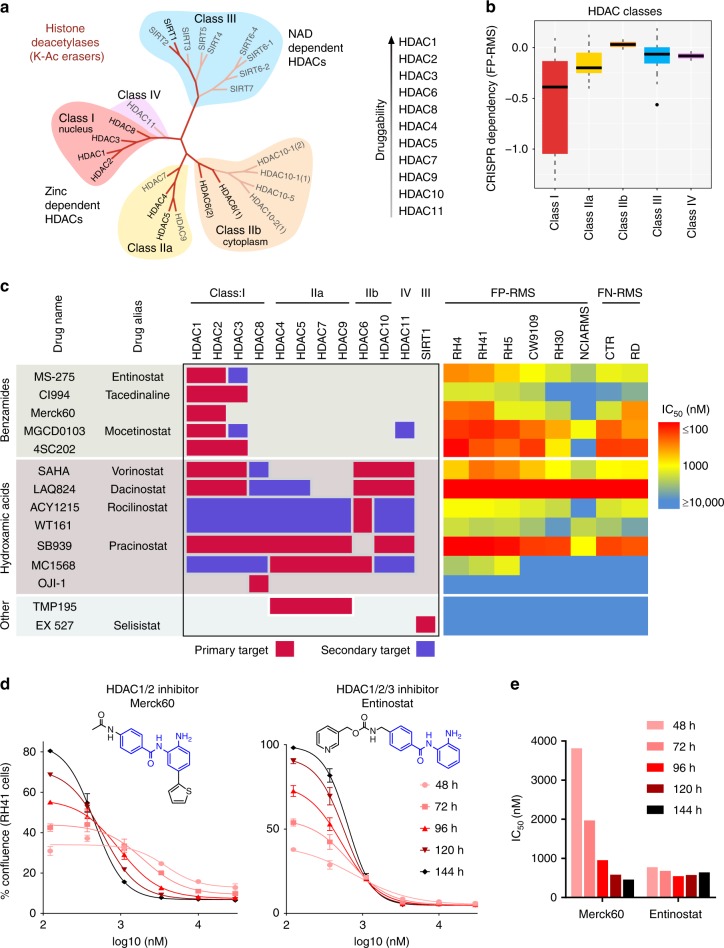
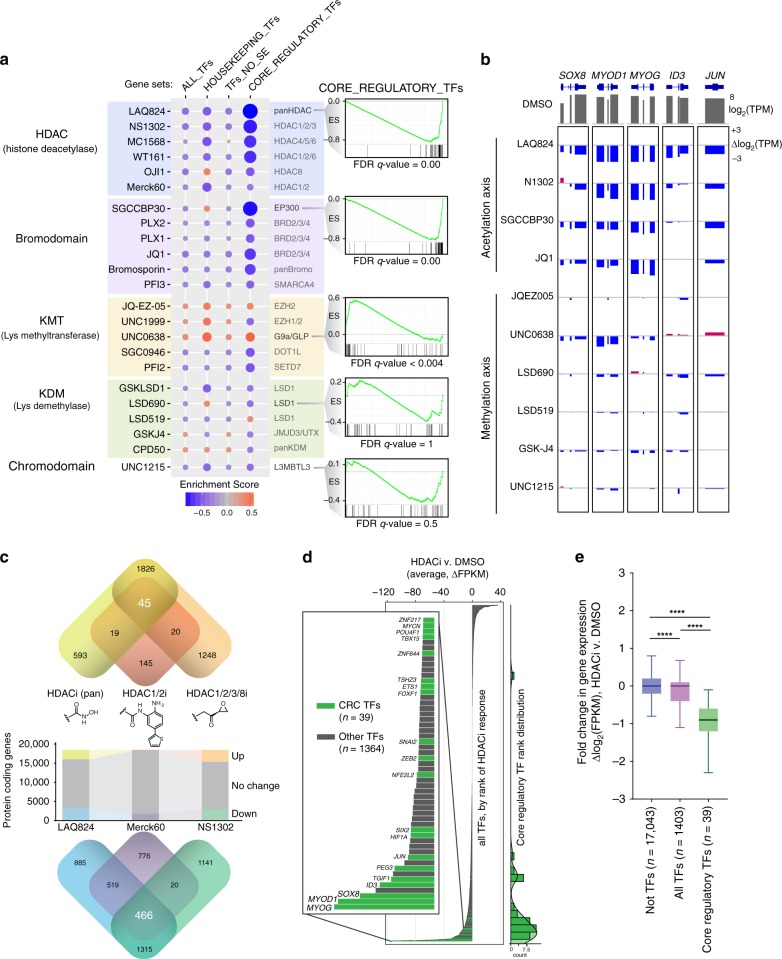
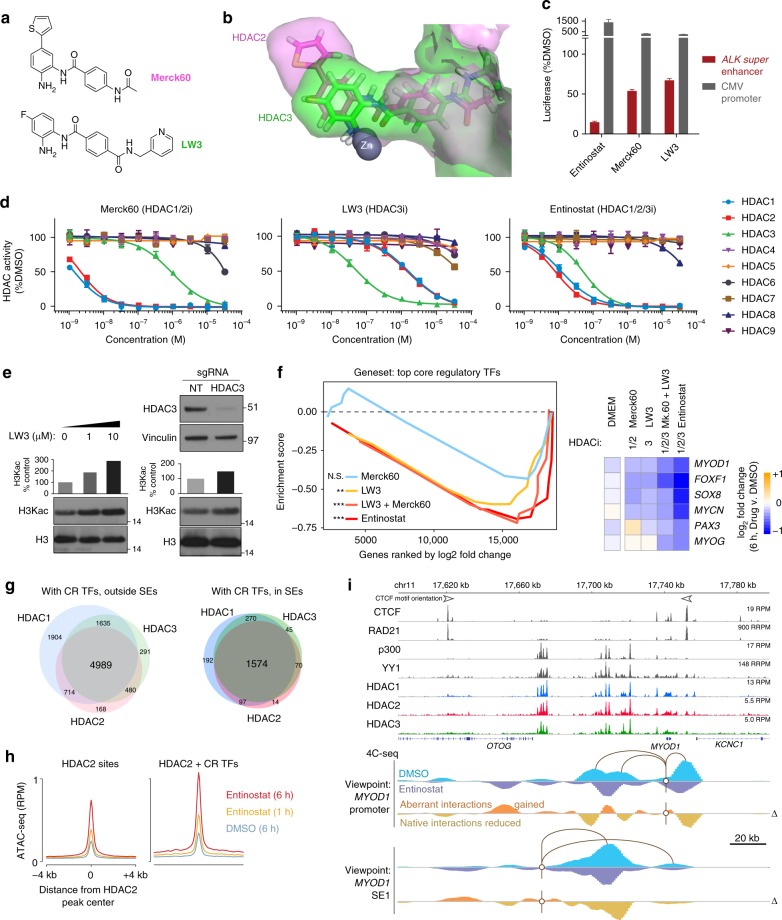
Identity determining transcription factors (TFs), or core regulatory (CR) TFs, are governed by cell-type specific super enhancers (SEs). Drugs to selectively inhibit CR circuitry are of high interest for cancer treatment. In alveolar rhabdomyosarcoma, PAX3-FOXO1 activates SEs to induce the expression of other CR TFs, providing a model system for studying cancer cell addiction to CR transcription. Using chemical genetics, the systematic screening of chemical matter for a biological outcome, here we report on a screen for epigenetic chemical probes able to distinguish between SE-driven transcription and constitutive transcription. We find that chemical probes along the acetylation-axis, and not the methylation-axis, selectively disrupt CR transcription. Additionally, we find that histone deacetylases (HDACs) are essential for CR TF transcription. We further dissect the contribution of HDAC isoforms using selective inhibitors, including the newly developed selective HDAC3 inhibitor LW3. We show HDAC1/2/3 are the co-essential isoforms that when co-inhibited halt CR transcription, making CR TF sites hyper-accessible and disrupting chromatin looping.
Conflict of interest statement
All authors declare no competing interests.
Figures

References
-
- Puissant A, et al. Targeting MYCN in neuroblastoma by BET bromodomain inhibition. Cancer Discov. 2013;3:308–323. doi: 10.1158/2159-8290.CD-12-0418. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- P01 CA066996/CA/NCI NIH HHS/United States
- P01 CA142106/CA/NCI NIH HHS/United States
- P01-CA066996/U.S. Department of Health & Human Services | NIH | National Cancer Institute (NCI)/International
- P01-CA142106/U.S. Department of Health & Human Services | NIH | National Cancer Institute (NCI)/International
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
