

Report on three additional patients and genotype-phenotype correlation in SLC25A22-related disorders group
- PMID: 31285529
- PMCID: PMC6871179
- DOI: 10.1038/s41431-019-0433-2
Report on three additional patients and genotype-phenotype correlation in SLC25A22-related disorders group
Abstract
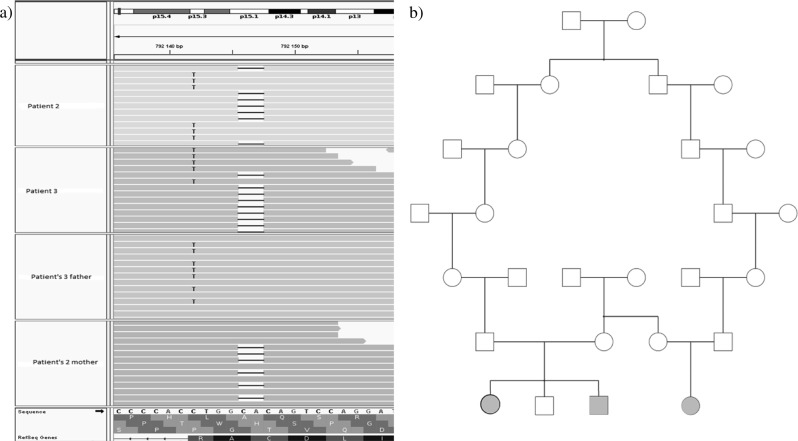
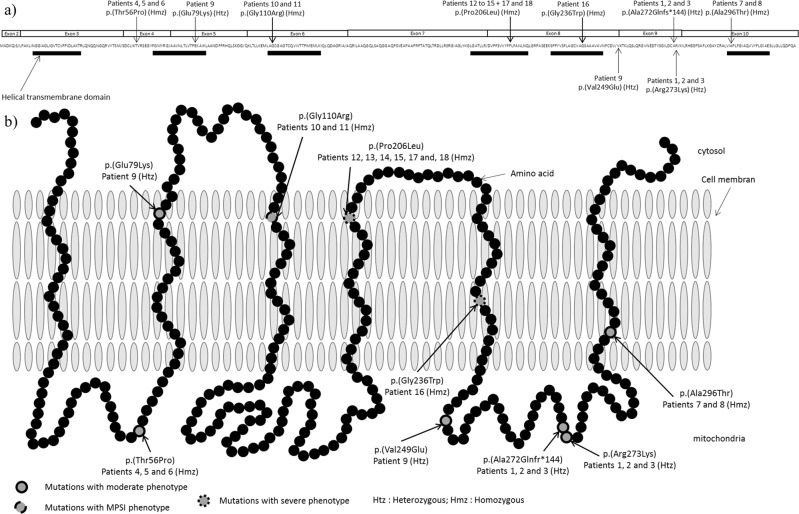
Early infantile epileptic encephalopathy (EIEE) is a heterogeneous group of severe forms of age-related developmental and epileptic encephalopathies with onset during the first weeks or months of life. The interictal electroencephalogram (EEG) shows a "suppression burst" (SB) pattern. The prognosis is usually poor and most children die within the first two years or survive with very severe intellectual disabilities. EIEE type 3 is caused by variants affecting function, in SLC25A22, which is also responsible for epilepsy of infancy with migrating focal seizures (EIMFS). We report a family with a less severe phenotype of EIEE type 3. We performed exome sequencing and identified two unreported variants in SLC25A22 in the compound heterozygous state: NM_024698.4: c.[813_814delTG];[818 G>A] (p.[Ala272Glnfs*144];[Arg273Lys]). Functional studies in cultured skin fibroblasts from a patient showed that glutamate oxidation was strongly defective, based on a literature review. We clustered the 18 published patients (including those from this family) into three groups according to the severity of the SLC25A22-related disorders. In an attempt to identify genotype-phenotype correlations, we compared the variants according to the location depending on the protein domains. We observed that patients with two variants located in helical transmembrane domains presented a severe phenotype, whereas patients with at least one variant outside helical transmembrane domains presented a milder phenotype. These data are suggestive of a continuum of disorders related to SLC25A22 that could be called SLC25A22-related disorders. This might be a first clue to enable geneticists to outline a prognosis based on genetic molecular data regarding the SLC25A22 gene.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures

Similar articles
-
The phenotype caused by recessive variations in SLC25A22: Report of a new case and literature review.Arch Pediatr. 2021 Jan;28(1):87-92. doi: 10.1016/j.arcped.2020.10.015. Epub 2020 Dec 17. Arch Pediatr. 2021. PMID: 33342683 Review.
-
Severe early-onset developmental and epileptic encephalopathy (DEE) associated with novel compound heterozygous pathogenic variants in SLC25A22: Case report and literature review.Seizure. 2019 Aug;70:56-58. doi: 10.1016/j.seizure.2019.06.029. Epub 2019 Jun 27. Seizure. 2019. PMID: 31279168 Review. No abstract available.
-
SLC25A22 is a novel gene for migrating partial seizures in infancy.Ann Neurol. 2013 Dec;74(6):873-82. doi: 10.1002/ana.23998. Ann Neurol. 2013. PMID: 24596948 Free PMC article.
-
De novo mutation in SLC25A22 gene: expansion of the clinical and electroencephalographic phenotype.J Neurogenet. 2021 Mar-Jun;35(2):67-73. doi: 10.1080/01677063.2021.1892094. Epub 2021 Apr 6. J Neurogenet. 2021. PMID: 33821742
-
Two siblings with early infantile myoclonic encephalopathy due to mutation in the gene encoding mitochondrial glutamate/H+ symporter SLC25A22.Eur J Paediatr Neurol. 2014 Nov;18(6):801-5. doi: 10.1016/j.ejpn.2014.06.007. Epub 2014 Jul 5. Eur J Paediatr Neurol. 2014. PMID: 25033742
Cited by
-
Diseases Caused by Mutations in Mitochondrial Carrier Genes SLC25: A Review.Biomolecules. 2020 Apr 23;10(4):655. doi: 10.3390/biom10040655. Biomolecules. 2020. PMID: 32340404 Free PMC article. Review.
-
An integrated bioinformatic investigation of mitochondrial solute carrier family 25 (SLC25) in colon cancer followed by preliminary validation of member 5 (SLC25A5) in tumorigenesis.Cell Death Dis. 2022 Mar 14;13(3):237. doi: 10.1038/s41419-022-04692-1. Cell Death Dis. 2022. PMID: 35288533 Free PMC article.
-
The SLC25 Carrier Family: Important Transport Proteins in Mitochondrial Physiology and Pathology.Physiology (Bethesda). 2020 Sep 1;35(5):302-327. doi: 10.1152/physiol.00009.2020. Physiology (Bethesda). 2020. PMID: 32783608 Free PMC article. Review.
-
Amino Acid Transport Defects in Human Inherited Metabolic Disorders.Int J Mol Sci. 2019 Dec 23;21(1):119. doi: 10.3390/ijms21010119. Int J Mol Sci. 2019. PMID: 31878022 Free PMC article. Review.
-
Mitochondrial Carriers for Aspartate, Glutamate and Other Amino Acids: A Review.Int J Mol Sci. 2019 Sep 10;20(18):4456. doi: 10.3390/ijms20184456. Int J Mol Sci. 2019. PMID: 31510000 Free PMC article. Review.
References
-
- Fiermonte G, Palmieri L, Todisco S, Agrimi G, Palmieri F, Walker JE. Identification of the mitochondrial glutamate transporter. Bacterial expression, reconstitution, functional characterization, and tissue distribution of two human isoforms. J Biol Chem. 2002;277:19289–94. doi: 10.1074/jbc.M201572200. - DOI - PubMed
-
- Cohen R, Basel-Vanagaite L, Goldberg-Stern H, Halevy A, Shuper A, Feingold-Zadok M, et al. Two siblings with early infantile myoclonic encephalopathy due to mutation in the gene encoding mitochondrial glutamate/H+ symporter SLC25A22. Eur J Paediatr Neurol. 2014;18:801–5. doi: 10.1016/j.ejpn.2014.06.007. - DOI - PubMed
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
