

E1A oncogene induced sensitization to NK cell induced apoptosis requires PIDD and Caspase-2
- PMID: 31285853
- PMCID: PMC6602934
- DOI: 10.1038/s41420-019-0189-z
E1A oncogene induced sensitization to NK cell induced apoptosis requires PIDD and Caspase-2
Abstract
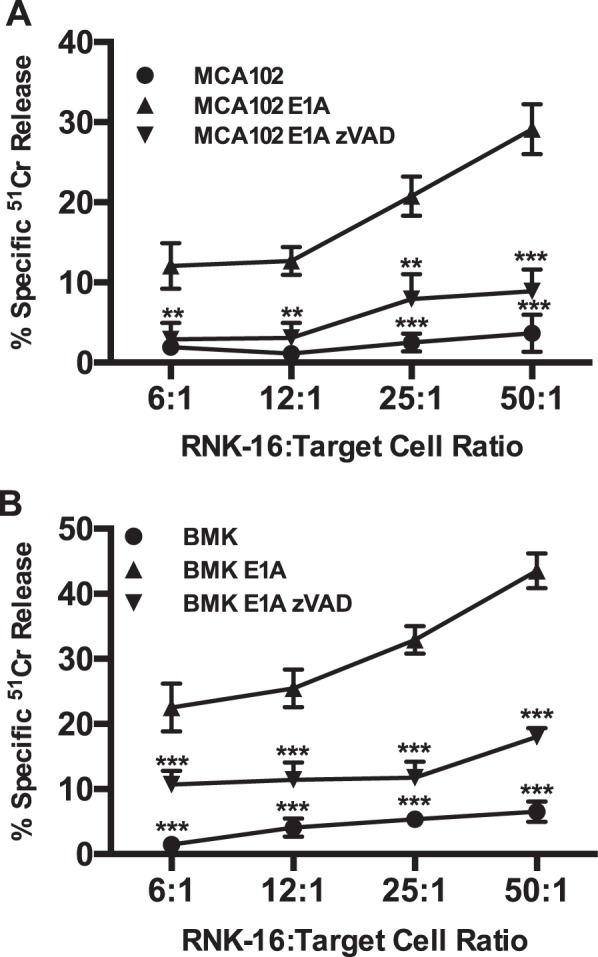
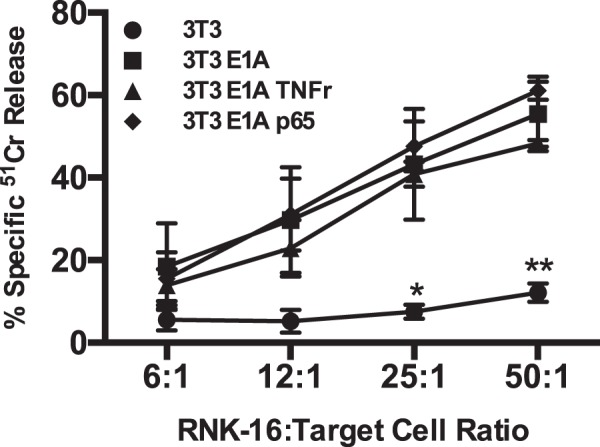
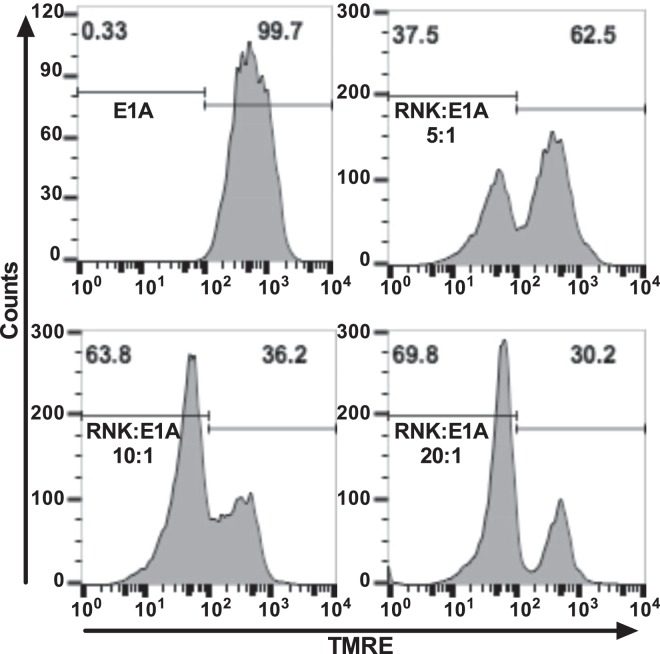
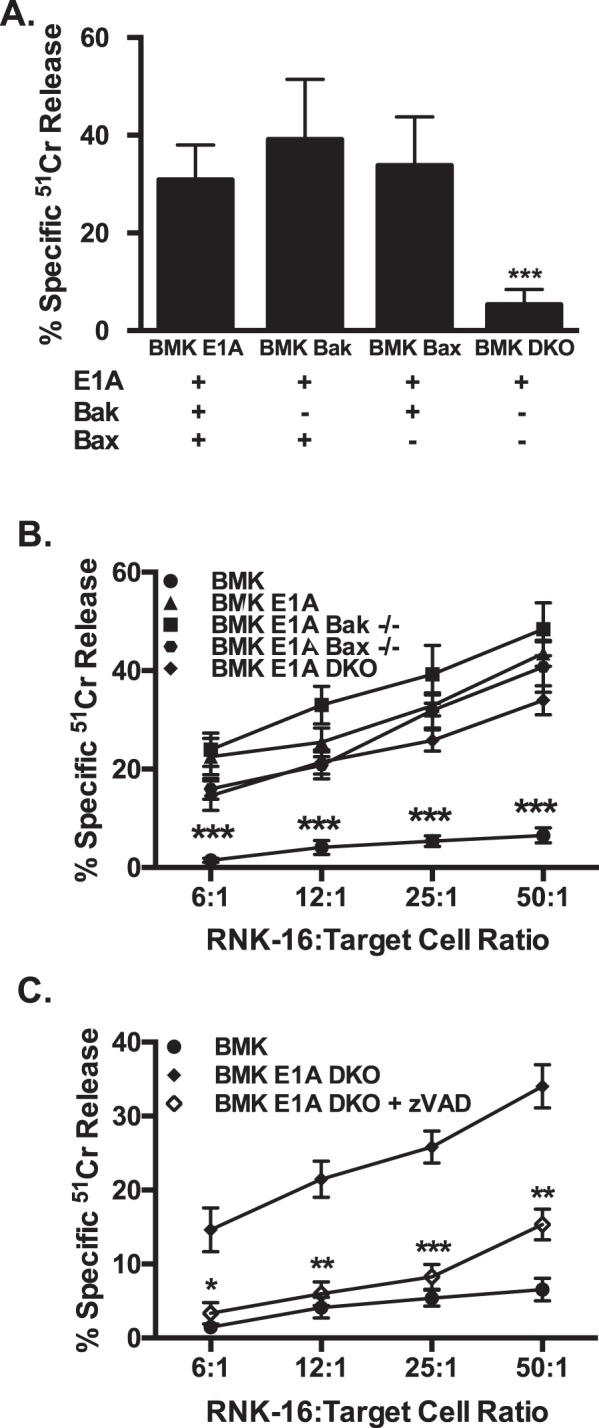
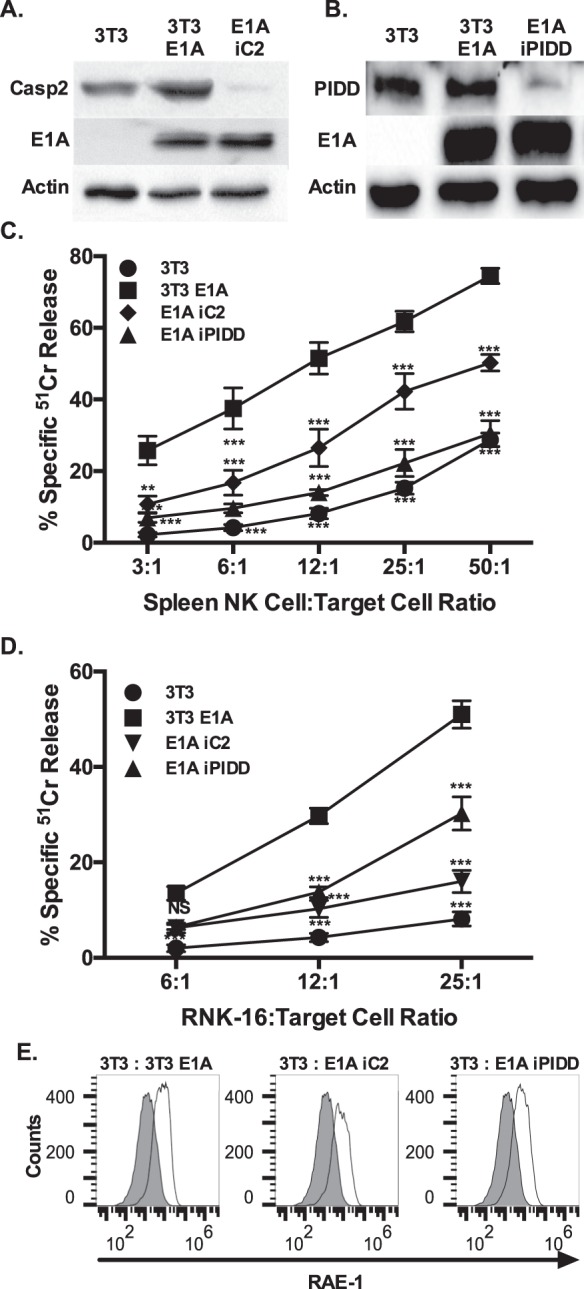
Expression of the adenovirus E1A oncogene sensitizes tumor cells to innate immune rejection by NK cells. This increased NK sensitivity is only partly explained by an E1A-induced increase in target cell surface expression of NKG2D ligands. The post-recognition mechanisms by which E1A sensitizes cells to the apoptotic cell death response to NK injury remains to be defined. E1A sensitizes cells to apoptotic stimuli through two distinct mechanisms-repression of NF-κB-dependent antiapoptotic responses and enhancement of caspase-2 activation and related mitochondrial injury. The current studies examined the roles of each of these post-NKG2D-recognition pathways in the increased sensitivity of E1A-positive target cells to NK killing. Sensitization to NK-induced apoptosis was independent of E1A-mediated repression of cellular NF-κB responses but was dependent on the expression of both caspase-2 and the upstream, caspase-2 activating molecule, PIDD. Target cells lacking caspase-2 or PIDD expression retained E1A-induced increased expression of the NKG2D ligand, RAE-1. NK cell-induced mitochondrial injury of E1A-expressing cells did not require expression of the mitochondrial molecules, Bak or Bax. These results define a PIDD/caspase-2-dependent pathway, through which E1A sensitizes cells to NK-mediated cytolysis independently of and complementarily to E1A-enhanced NKG2D/RAE-1 ligand expression.
Keywords: Apoptosis; Innate immunity.
Conflict of interest statement
Conflict of interestThe authors declare that they have no conflict of interest.
Figures
References
-
- Cook, J. L. & Routes, J. M. Role of the innate immune response in determining the tumorigenicity of neoplastic cells. 106, 99–107; discussion 107–108, 143–160 (2001). - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
