

New insights into the interplay between autophagy, gut microbiota and inflammatory responses in IBD
- PMID: 31286804
- PMCID: PMC6984609
- DOI: 10.1080/15548627.2019.1635384
New insights into the interplay between autophagy, gut microbiota and inflammatory responses in IBD
Abstract
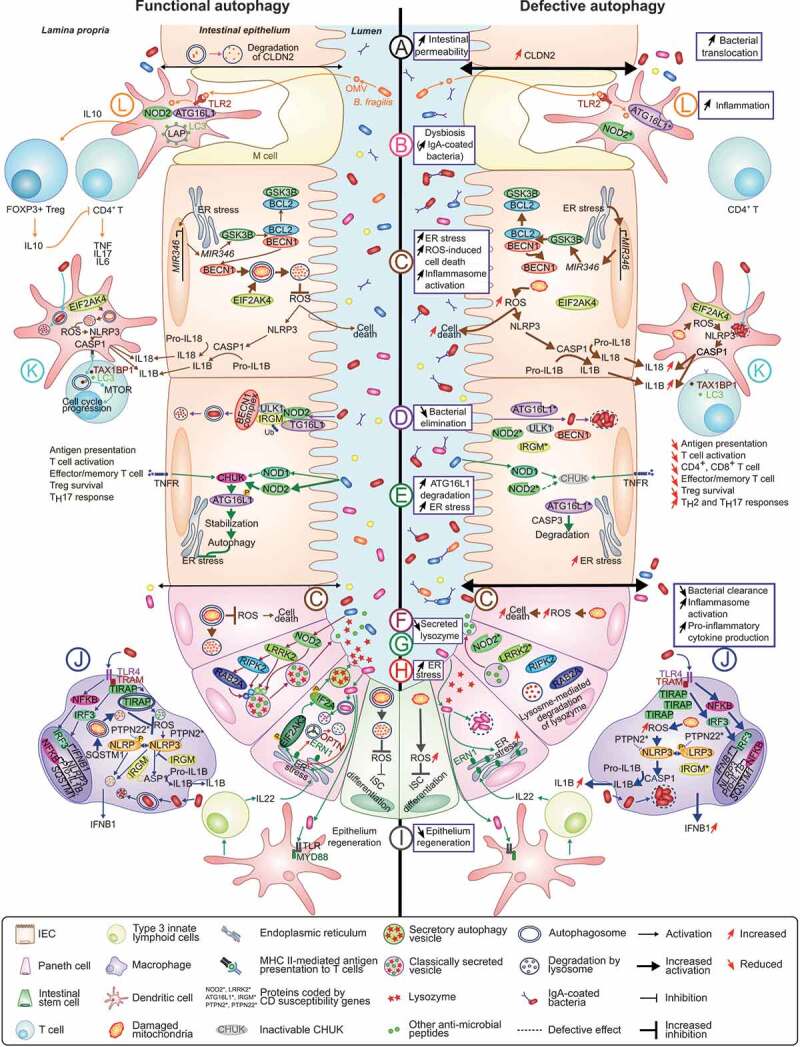
One of the most significant challenges of inflammatory bowel disease (IBD) research is to understand how alterations in the symbiotic relationship between the genetic composition of the host and the intestinal microbiota, under impact of specific environmental factors, lead to chronic intestinal inflammation. Genome-wide association studies, followed by functional studies, have identified a role for numerous autophagy genes in IBD, especially in Crohn disease. Studies using in vitro and in vivo models, in addition to human clinical studies have revealed that autophagy is pivotal for intestinal homeostasis maintenance, gut ecology regulation, appropriate intestinal immune responses and anti-microbial protection. This review describes the latest researches on the mechanisms by which dysfunctional autophagy leads to disrupted intestinal epithelial function, gut dysbiosis, defect in anti-microbial peptide secretion by Paneth cells, endoplasmic reticulum stress response and aberrant immune responses to pathogenic bacteria. A better understanding of the role of autophagy in IBD pathogenesis may provide better sub-classification of IBD phenotypes and novel approaches for disease management.Abbreviations: AIEC: adherent-invasive Escherichia coli; AMPK: AMP-activated protein kinase; ATF6: activating transcription factor 6; ATG: autophagy related; Atg16l1[ΔIEC] mice: mice with Atg16l1 depletion specifically in intestinal epithelial cells; Atg16l1[HM] mice: mice hypomorphic for Atg16l1 expression; BCL2: B cell leukemia/lymphoma 2; BECN1: beclin 1, autophagy related; CALCOCO2: calcium binding and coiled-coil domain 2; CASP: caspase; CD: Crohn disease; CGAS: cyclic GMP-AMP synthase; CHUK/IKKA: conserved helix-loop-helix ubiquitous kinase; CLDN2: claudin 2; DAPK1: death associated protein kinase 1; DCs: dendritic cells; DSS: dextran sulfate sodium; EIF2A: eukaryotic translation initiation factor 2A; EIF2AK: eukaryotic translation initiation factor 2 alpha kinase; ER: endoplasmic reticulum; ERBIN: Erbb2 interacting protein; ERN1/IRE1A: ER to nucleus signaling 1; FNBP1L: formin binding protein 1-like; FOXP3: forkhead box P3; GPR65: G-protein coupled receptor 65; GSK3B: glycogen synthase kinase 3 beta; IBD: inflammatory bowel disease; IECs: intestinal epithelial cells; IFN: interferon; IL: interleukin; IL10R: interleukin 10 receptor; IRGM: immunity related GTPase M; ISC: intestinal stem cell; LAMP1: lysosomal-associated membrane protein 1; LAP: LC3-associated phagocytosis; MAP1LC3B: microtubule-associated protein 1 light chain 3 beta; LPS: lipopolysaccharide; LRRK2: leucine-rich repeat kinase 2; MAPK: mitogen-activated protein kinase; MHC: major histocompatibility complex; MIF: macrophage migration inhibitory factor; MIR/miRNA: microRNA; MTMR3: myotubularin related protein 3; MTOR: mechanistic target of rapamycin kinase; MYD88: myeloid differentiation primary response gene 88; NLRP3: NLR family, pyrin domain containing 3; NOD2: nucleotide-binding oligomerization domain containing 2; NPC: Niemann-Pick disease type C; NPC1: NPC intracellular cholesterol transporter 1; OMVs: outer membrane vesicles; OPTN: optineurin; PI3K: phosphoinositide 3-kinase; PRR: pattern-recognition receptor; PTPN2: protein tyrosine phosphatase, non-receptor type 2; PTPN22: protein tyrosine phosphatase, non-receptor type 22 (lymphoid); PYCARD/ASC: PYD and CARD domain containing; RAB2A: RAB2A, member RAS oncogene family; RELA: v-rel reticuloendotheliosis viral oncogene homolog A (avian); RIPK2: receptor (TNFRSF)-interacting serine-threonine kinase 2; ROS: reactive oxygen species; SNPs: single nucleotide polymorphisms; SQSTM1: sequestosome 1; TAX1BP1: Tax1 binding protein 1; Th: T helper 1; TIRAP/TRIF: toll-interleukin 1 receptor (TIR) domain-containing adaptor protein; TLR: toll-like receptor; TMEM173/STING: transmembrane protein 173; TMEM59: transmembrane protein 59; TNF/TNFA: tumor necrosis factor; Treg: regulatory T; TREM1: triggering receptor expressed on myeloid cells 1; UC: ulcerative colitis; ULK1: unc-51 like autophagy activating kinase 1; WT: wild-type; XBP1: X-box binding protein 1; XIAP: X-linked inhibitor of apoptosis.
Keywords: Autophagy; immune responses; inflammatory bowel diseases; intestinal homeostasis; intestinal microbiota; microbial infection.
Figures
References
-
- Carrière J, Darfeuille-Michaud A, Nguyen HTT.. Infectious etiopathogenesis of crohn’s disease. World J Gastroenterol. [Internet] 2014;20:12102–12117. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25232246 - PMC - PubMed
-
- Nguyen HTT, Lapaquette P, Bringer M-A, et al. Autophagy and crohn’s disease. J Innate Immun. [Internet] 2013;5:434–443. Available from: https://www.karger.com/Article/FullText/345129 - PMC - PubMed
-
- Mizushima N. A brief history of autophagy from cell biology to physiology and disease [Internet]. Nat Cell Bio. 2018;20:521–527. Available from: http://www.nature.com/articles/s41556-018-0092-5 - PubMed
-
- Kim S, Eun H, Jo E-K. Roles of autophagy-related genes in the pathogenesis of inflammatory bowel disease. Cells. [Internet] 2019;8:77. Available from: http://www.mdpi.com/2073-4409/8/1/77 - PMC - PubMed
-
- Lapaquette P, Thi Thu Nguyen H, Faure M. L’autophagie garante de l’immunité et de l’inflammation. médecine/sciences. [Internet] 2017;33:305–311. Available from: http://www.medecinesciences.org/10.1051/medsci/20173303018 - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous