

Autophagy regulates inflammatory programmed cell death via turnover of RHIM-domain proteins
- PMID: 31287416
- PMCID: PMC6615860
- DOI: 10.7554/eLife.44452
Autophagy regulates inflammatory programmed cell death via turnover of RHIM-domain proteins
Abstract
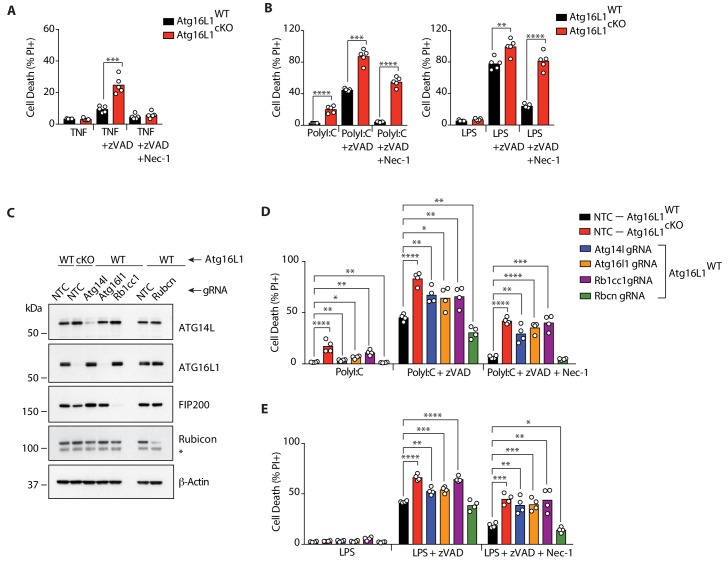
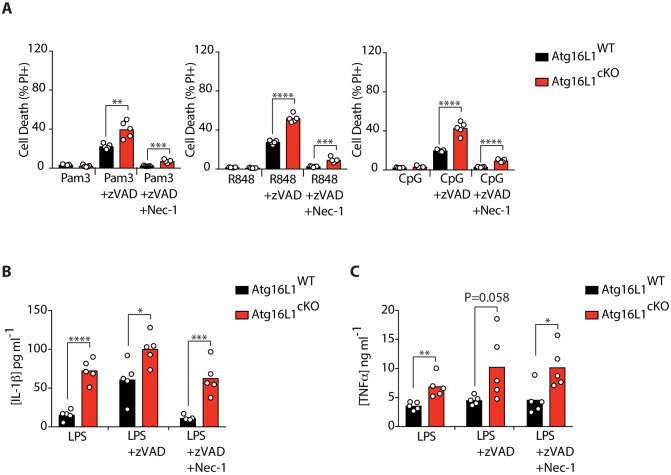
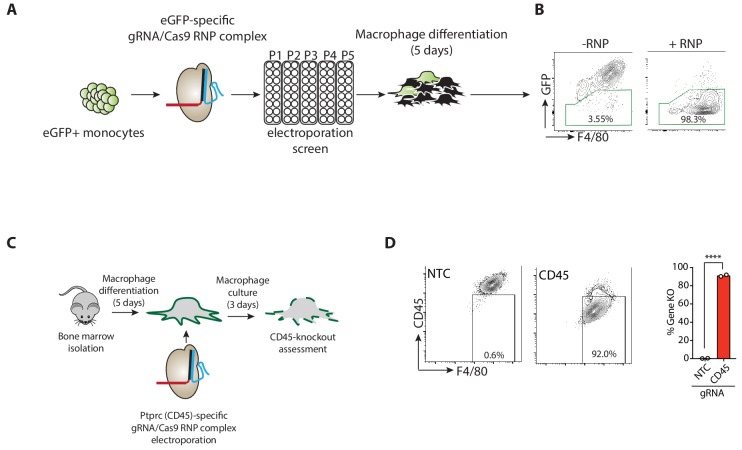
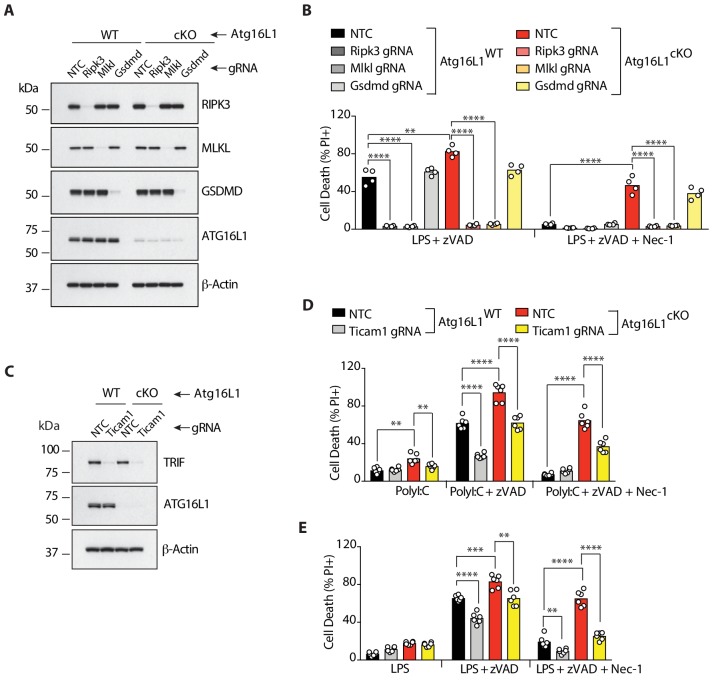
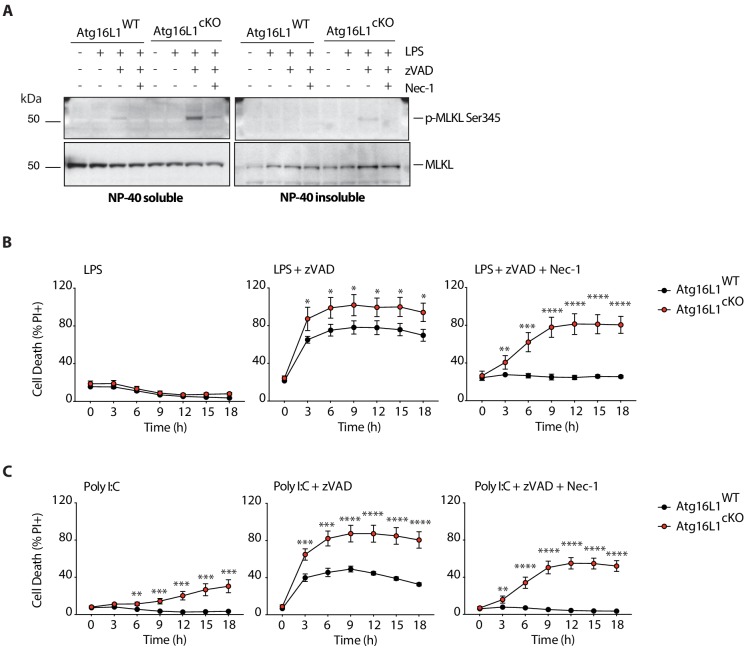
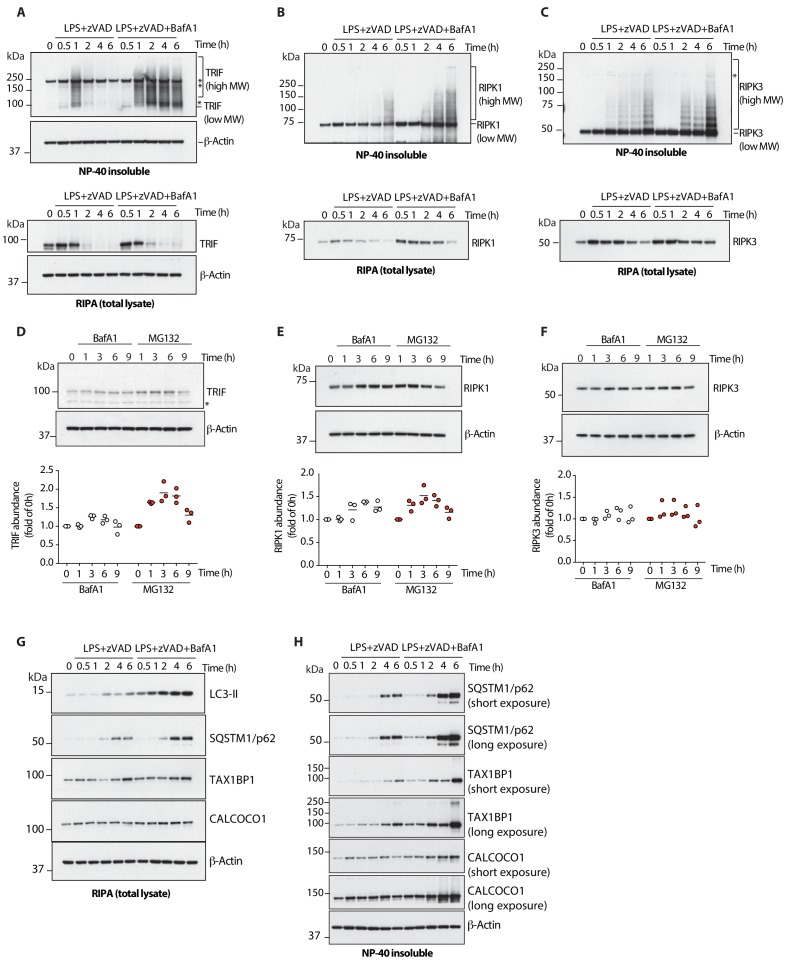
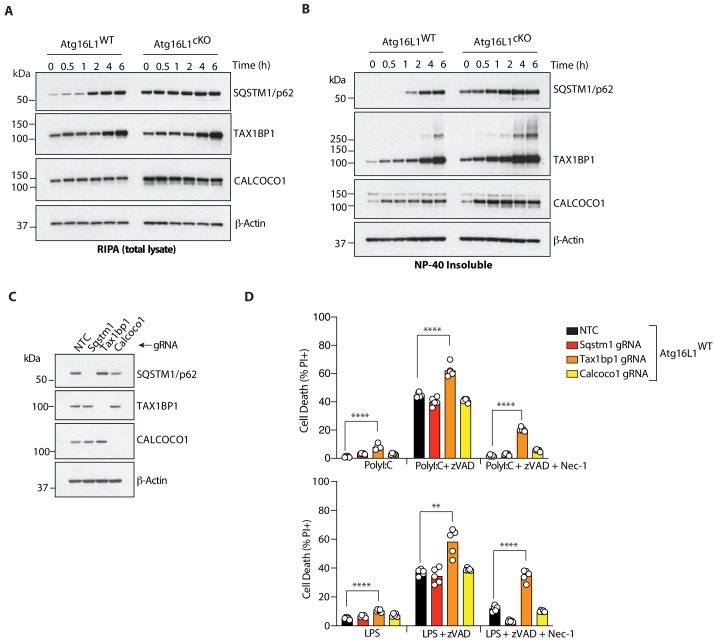
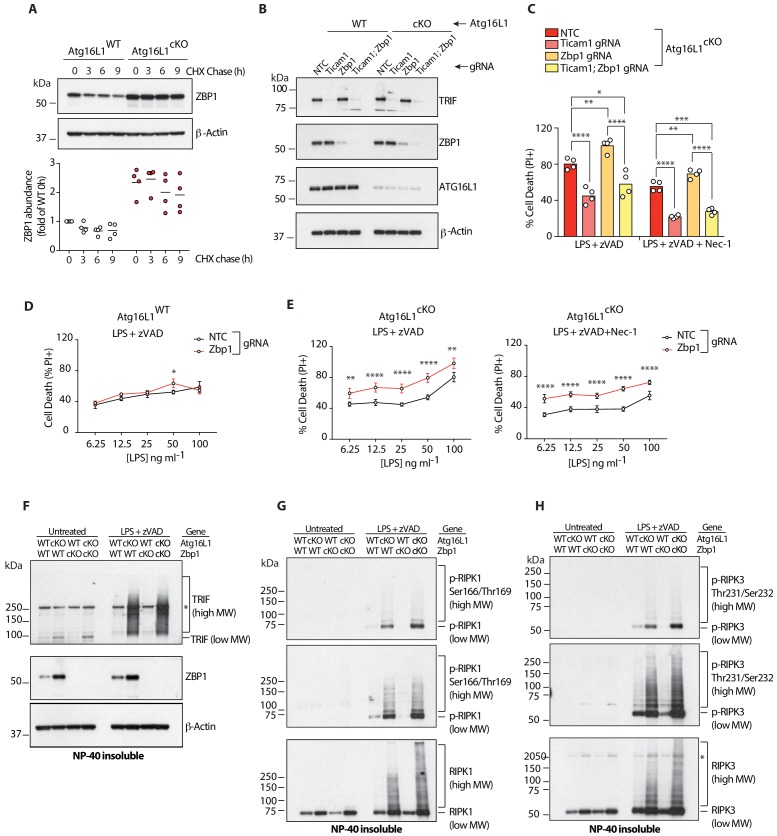
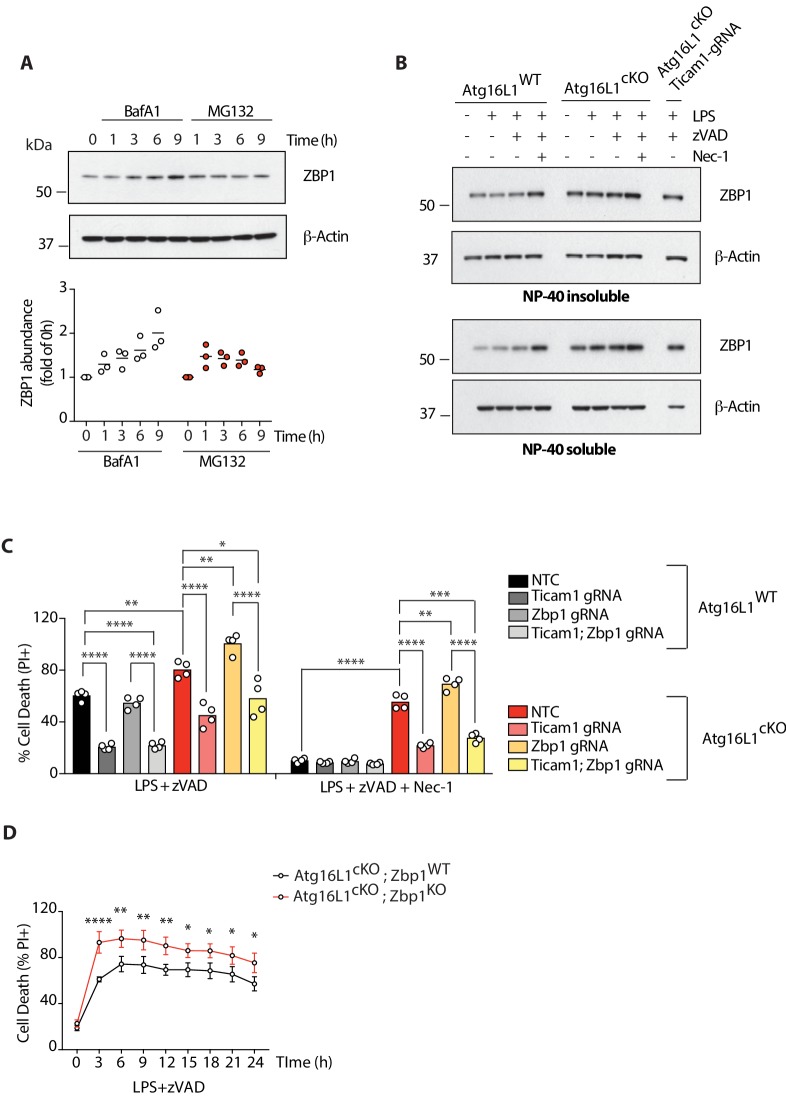
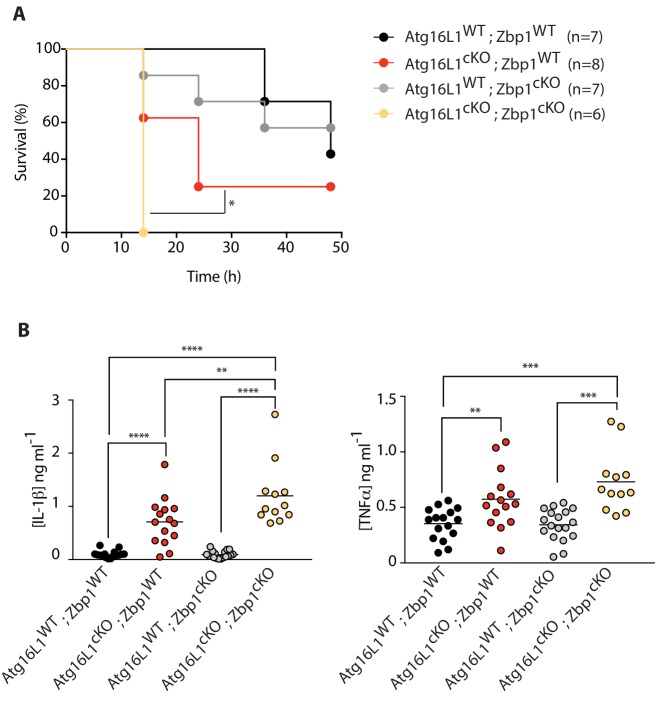
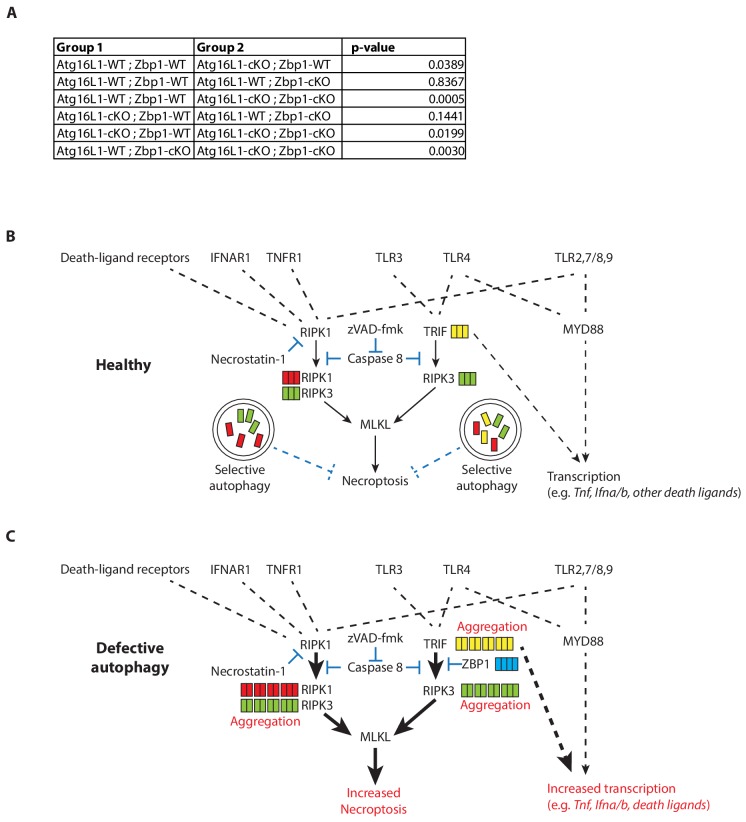
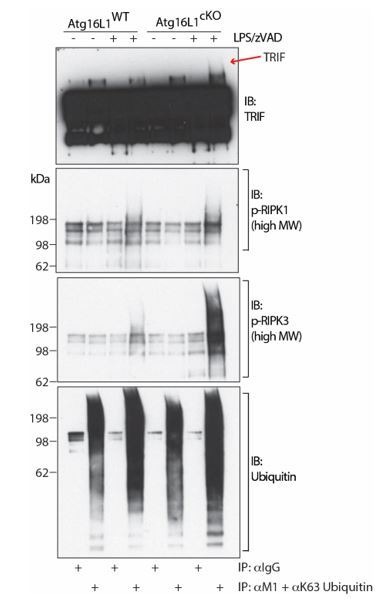
RIPK1, RIPK3, ZBP1 and TRIF, the four mammalian proteins harboring RIP homotypic interaction motif (RHIM) domains, are key components of inflammatory signaling and programmed cell death. RHIM-domain protein activation is mediated by their oligomerization; however, mechanisms that promote a return to homeostasis remain unknown. Here we show that autophagy is critical for the turnover of all RHIM-domain proteins. Macrophages lacking the autophagy gene Atg16l1accumulated highly insoluble forms of RIPK1, RIPK3, TRIF and ZBP1. Defective autophagy enhanced necroptosis by Tumor necrosis factor (TNF) and Toll-like receptor (TLR) ligands. TNF-mediated necroptosis was mediated by RIPK1 kinase activity, whereas TLR3- or TLR4-mediated death was dependent on TRIF and RIPK3. Unexpectedly, combined deletion of Atg16l1 and Zbp1 accelerated LPS-mediated necroptosis and sepsis in mice. Thus, ZBP1 drives necroptosis in the absence of the RIPK1-RHIM, but suppresses this process when multiple RHIM-domain containing proteins accumulate. These findings identify autophagy as a central regulator of innate inflammation governed by RHIM-domain proteins.
Keywords: CRISPR; autophagy; cell biology; immunology; inflammation; macrophage; mouse; necroptosis; signaling.
© 2019, Lim et al.
Conflict of interest statement
JL, HP, JH, TM, MR, MX, BM, Mv, KN, AM Affiliated with Genentech. No other competing interests to declare
Figures
Similar articles
-
RIPK1 inhibits ZBP1-driven necroptosis during development.Nature. 2016 Dec 1;540(7631):129-133. doi: 10.1038/nature20559. Epub 2016 Nov 7. Nature. 2016. PMID: 27819682
-
The RIPK1 death domain restrains ZBP1- and TRIF-mediated cell death and inflammation.Immunity. 2024 Jul 9;57(7):1497-1513.e6. doi: 10.1016/j.immuni.2024.04.016. Epub 2024 May 13. Immunity. 2024. PMID: 38744293
-
RIPK1 counteracts ZBP1-mediated necroptosis to inhibit inflammation.Nature. 2016 Dec 1;540(7631):124-128. doi: 10.1038/nature20558. Epub 2016 Nov 7. Nature. 2016. PMID: 27819681 Free PMC article.
-
An outline of necrosome triggers.Cell Mol Life Sci. 2016 Jun;73(11-12):2137-52. doi: 10.1007/s00018-016-2189-y. Epub 2016 Apr 6. Cell Mol Life Sci. 2016. PMID: 27052312 Free PMC article. Review.
-
Initiation and execution mechanisms of necroptosis: an overview.Cell Death Differ. 2017 Jul;24(7):1184-1195. doi: 10.1038/cdd.2017.65. Epub 2017 May 12. Cell Death Differ. 2017. PMID: 28498367 Free PMC article. Review.
Cited by
-
Necroptosis as a Novel Facet of Mitotic Catastrophe.Int J Mol Sci. 2022 Mar 29;23(7):3733. doi: 10.3390/ijms23073733. Int J Mol Sci. 2022. PMID: 35409093 Free PMC article.
-
RIP3 in Necroptosis: Underlying Contributions to Traumatic Brain Injury.Neurochem Res. 2024 Feb;49(2):245-257. doi: 10.1007/s11064-023-04038-z. Epub 2023 Sep 25. Neurochem Res. 2024. PMID: 37743445 Review.
-
NLRP6 induces RIP1 kinase-dependent necroptosis via TAK1-mediated p38MAPK/MK2 phosphorylation in S. typhimurium infection.iScience. 2024 Feb 28;27(4):109339. doi: 10.1016/j.isci.2024.109339. eCollection 2024 Apr 19. iScience. 2024. PMID: 38500819 Free PMC article.
-
Lactobacillus (LA-1) and butyrate inhibit osteoarthritis by controlling autophagy and inflammatory cell death of chondrocytes.Front Immunol. 2022 Oct 17;13:930511. doi: 10.3389/fimmu.2022.930511. eCollection 2022. Front Immunol. 2022. PMID: 36325344 Free PMC article.
-
MLKL trafficking and accumulation at the plasma membrane control the kinetics and threshold for necroptosis.Nat Commun. 2020 Jun 19;11(1):3151. doi: 10.1038/s41467-020-16887-1. Nat Commun. 2020. PMID: 32561730 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
