

Genetic interactions of G-quadruplexes in humans
- PMID: 31287417
- PMCID: PMC6615864
- DOI: 10.7554/eLife.46793
Genetic interactions of G-quadruplexes in humans
Abstract
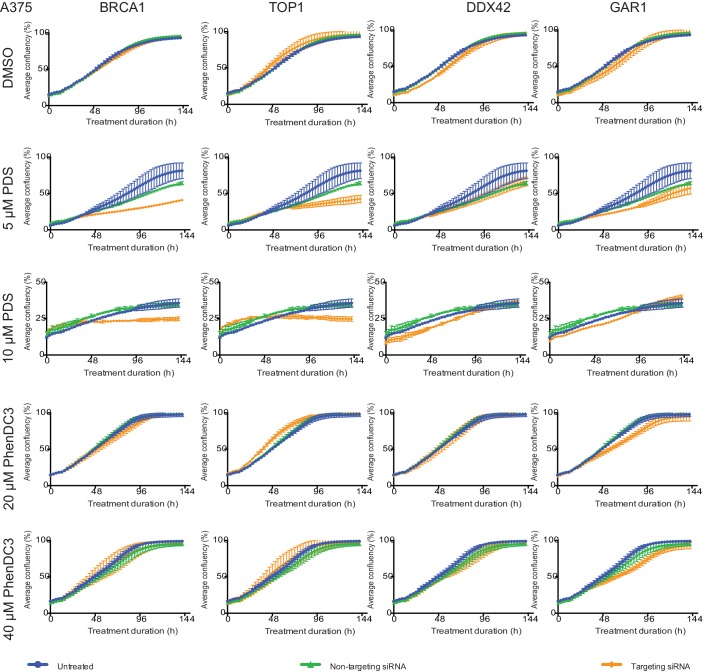
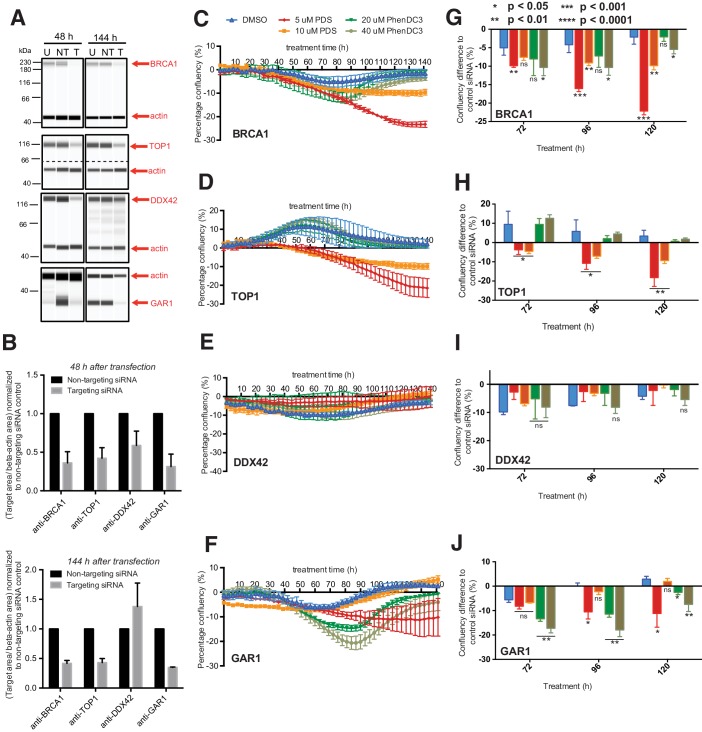
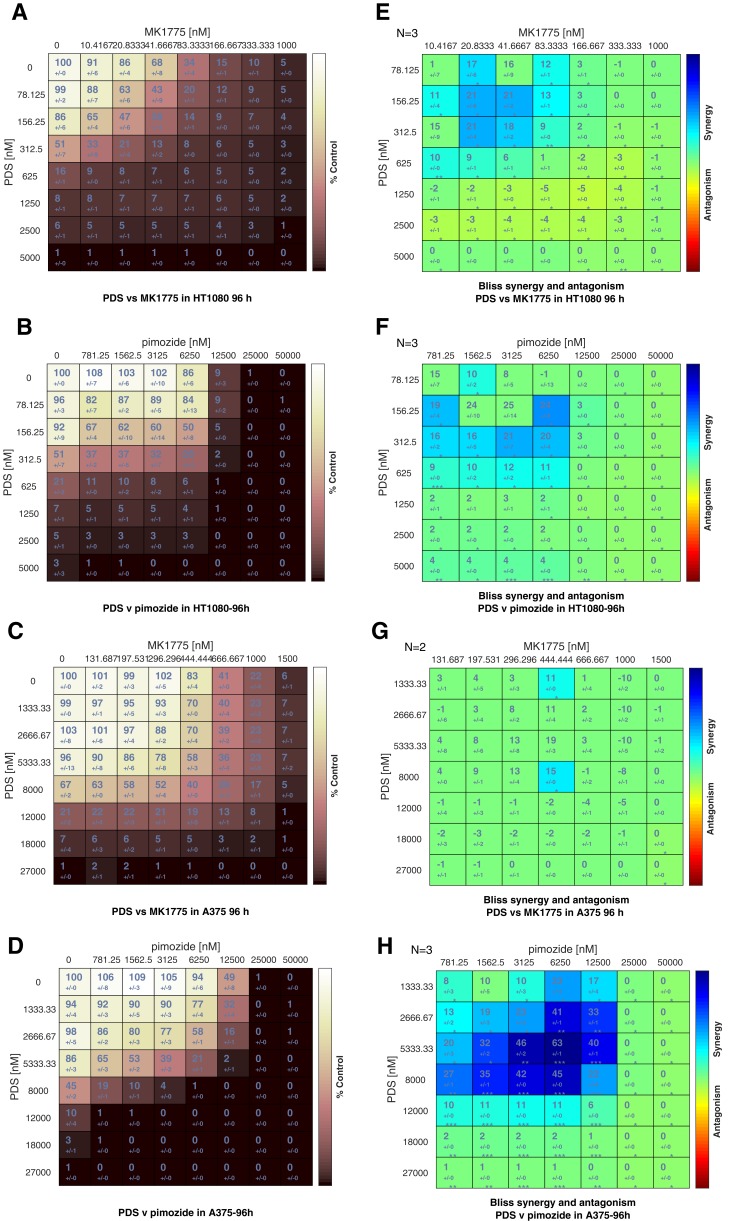
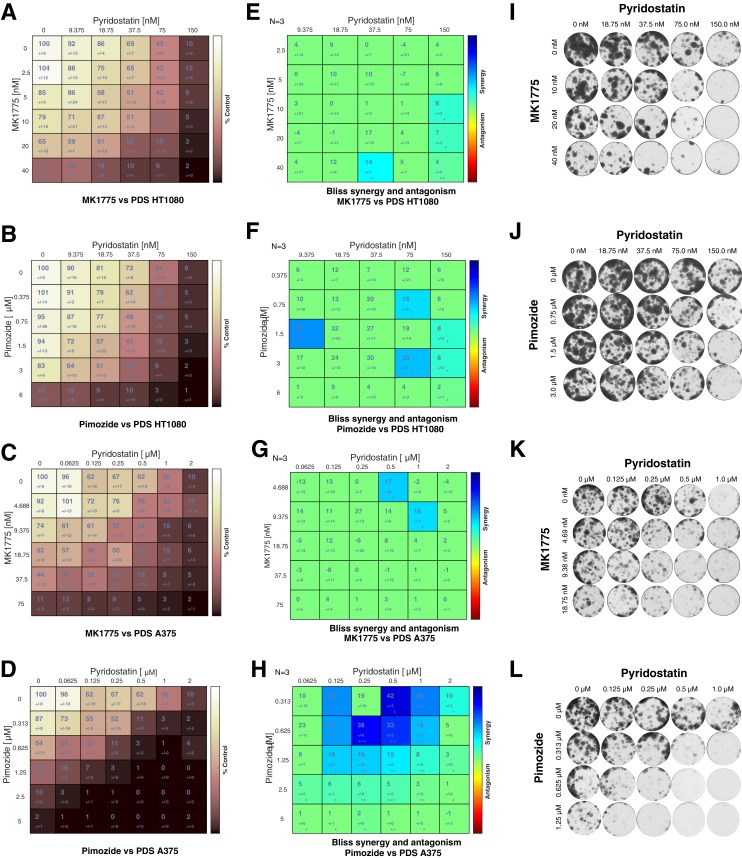
G-quadruplexes (G4) are alternative nucleic acid structures involved in transcription, translation and replication. Aberrant G4 formation and stabilisation is linked to genome instability and cancer. G4 ligand treatment disrupts key biological processes leading to cell death. To discover genes and pathways involved with G4s and gain mechanistic insights into G4 biology, we present the first unbiased genome-wide study to systematically identify human genes that promote cell death when silenced by shRNA in the presence of G4-stabilising small molecules. Many novel genetic vulnerabilities were revealed opening up new therapeutic possibilities in cancer, which we exemplified by an orthogonal pharmacological inhibition approach that phenocopies gene silencing. We find that targeting the WEE1 cell cycle kinase or USP1 deubiquitinase in combination with G4 ligand treatment enhances cell killing. We also identify new genes and pathways regulating or interacting with G4s and demonstrate that the DDX42 DEAD-box helicase is a newly discovered G4-binding protein.
Keywords: G-quadruplex; G-quadruplex ligands; G4; biochemistry; cancer; chemical biology; chromosomes; gene expression; genetic vulnerability; human; nucleic acid structure.
© 2019, Zyner et al.
Conflict of interest statement
KZ, DM, SA, SM, MD, NE, DT No competing interests declared, GH Is associated with transOMIC Technologies, who have commercialised libraries constructed using the shERWOOD and ultramiR design strategies. SB Is founder, adviser and shareholder of Cambridge Epigenetix Ltd.
Figures


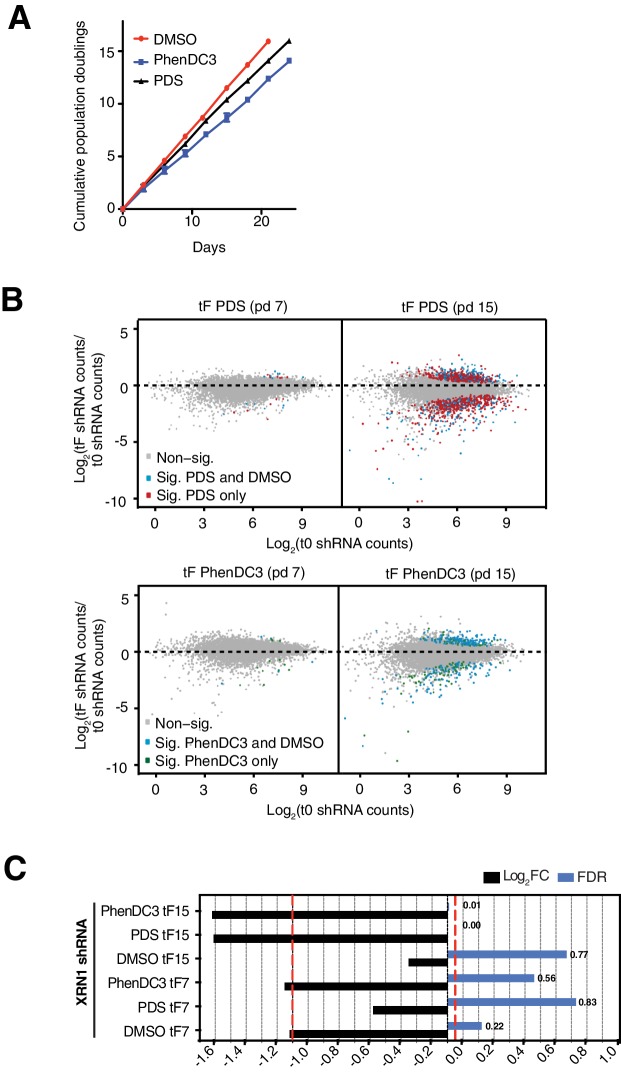




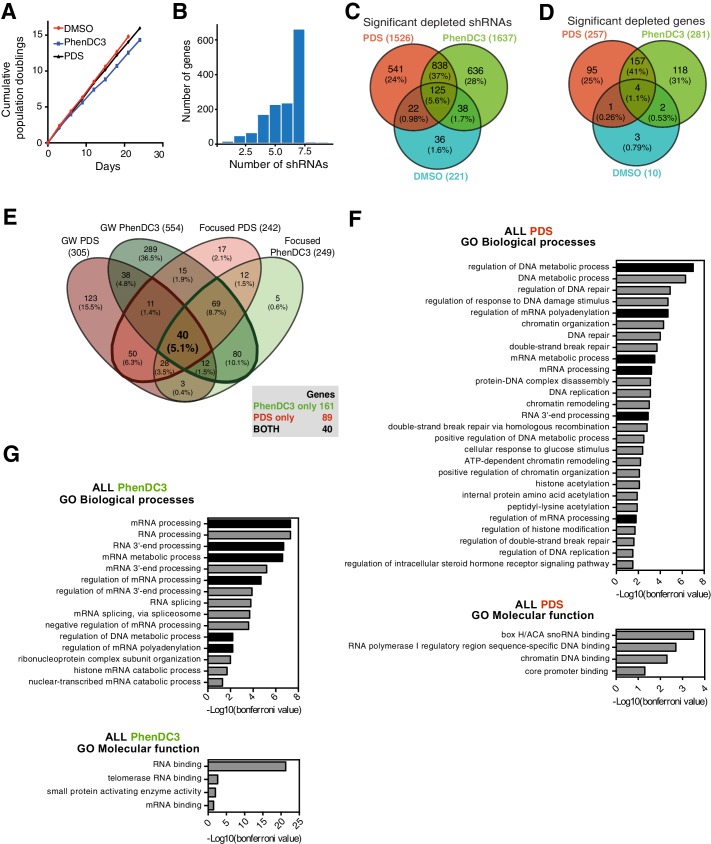

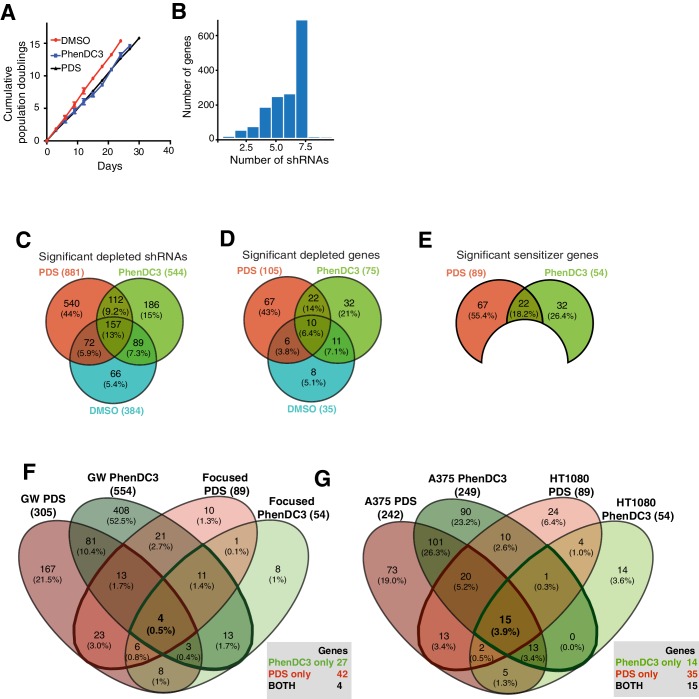

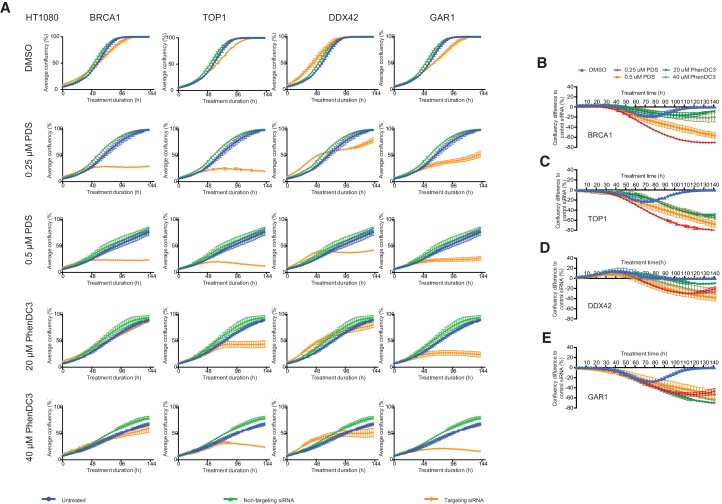



References
-
- Andrews S. Bioinformatics; 2010. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
-
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nature Genetics. 2000;25:25–34. doi: 10.1038/75556. - DOI - PMC - PubMed
-
- Azorsa DO, Gonzales IM, Basu GD, Choudhary A, Arora S, Bisanz KM, Kiefer JA, Henderson MC, Trent JM, Von Hoff DD, Mousses S. Synthetic lethal RNAi screening identifies sensitizing targets for gemcitabine therapy in pancreatic cancer. Journal of Translational Medicine. 2009;7:43. doi: 10.1186/1479-5876-7-43. - DOI - PMC - PubMed
-
- Backert S, Gelos M, Kobalz U, Hanski ML, Böhm C, Mann B, Lövin N, Gratchev A, Mansmann U, Moyer MP, Riecken EO, Hanski C. Differential gene expression in colon carcinoma cells and tissues detected with a cDNA array. International Journal of Cancer. 1999;82:868–874. doi: 10.1002/(SICI)1097-0215(19990909)82:6<868::AID-IJC16>3.0.CO;2-W. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
