

Managing genomic variant calling workflows with Swift/T
- PMID: 31287816
- PMCID: PMC6615596
- DOI: 10.1371/journal.pone.0211608
Managing genomic variant calling workflows with Swift/T
Abstract
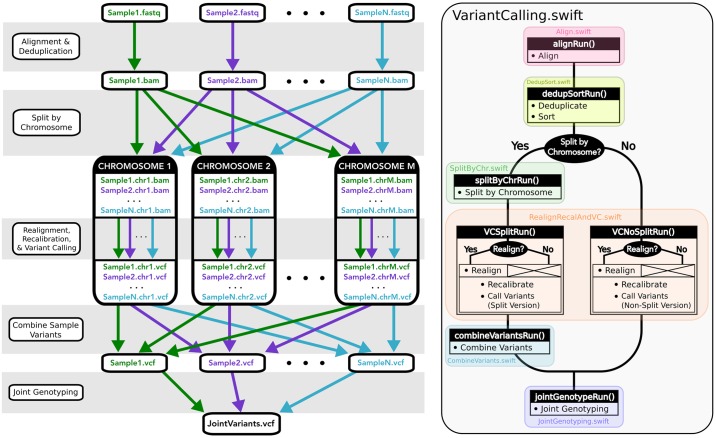
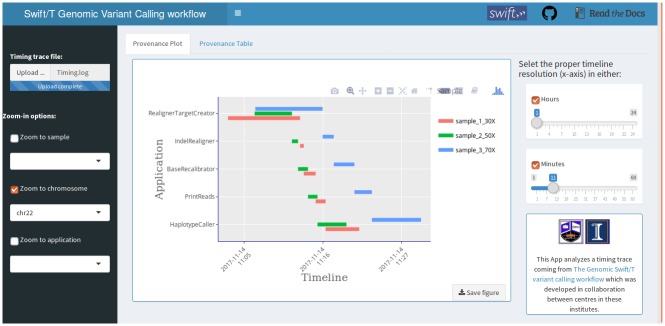
Bioinformatics research is frequently performed using complex workflows with multiple steps, fans, merges, and conditionals. This complexity makes management of the workflow difficult on a computer cluster, especially when running in parallel on large batches of data: hundreds or thousands of samples at a time. Scientific workflow management systems could help with that. Many are now being proposed, but is there yet the "best" workflow management system for bioinformatics? Such a system would need to satisfy numerous, sometimes conflicting requirements: from ease of use, to seamless deployment at peta- and exa-scale, and portability to the cloud. We evaluated Swift/T as a candidate for such role by implementing a primary genomic variant calling workflow in the Swift/T language, focusing on workflow management, performance and scalability issues that arise from production-grade big data genomic analyses. In the process we introduced novel features into the language, which are now part of its open repository. Additionally, we formalized a set of design criteria for quality, robust, maintainable workflows that must function at-scale in a production setting, such as a large genomic sequencing facility or a major hospital system. The use of Swift/T conveys two key advantages. (1) It operates transparently in multiple cluster scheduling environments (PBS Torque, SLURM, Cray aprun environment, etc.), thus a single workflow is trivially portable across numerous clusters. (2) The leaf functions of Swift/T permit developers to easily swap executables in and out of the workflow, which makes it easy to maintain and to request resources optimal for each stage of the pipeline. While Swift/T's data-level parallelism eliminates the need to code parallel analysis of multiple samples, it does make debugging more difficult, as is common for implicitly parallel code. Nonetheless, the language gives users a powerful and portable way to scale up analyses in many computing architectures. The code for our implementation of a variant calling workflow using Swift/T can be found on GitHub at https://github.com/ncsa/Swift-T-Variant-Calling, with full documentation provided at http://swift-t-variant-calling.readthedocs.io/en/latest/.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
