

Clinical features and genetic spectrum in Chinese patients with recessive hereditary spastic paraplegia
- PMID: 31289639
- PMCID: PMC6593507
- DOI: 10.1186/s40035-019-0157-9
Clinical features and genetic spectrum in Chinese patients with recessive hereditary spastic paraplegia
Abstract
Background: Although many causative genes of hereditary spastic paraplegia (HSP) have been uncovered in recent years, there are still approximately 50% of HSP patients without genetically diagnosis, especially in autosomal recessive (AR) HSP patients. Rare studies have been performed to determine the genetic spectrum and clinical profiles of recessive HSP patients in the Chinese population.
Methods: In this study, we investigated 24 Chinese index AR/sporadic patients by targeted next-generation sequencing (NGS), Sanger sequencing and multiplex ligation-dependent probe amplification (MLPA). Further functional studies were performed to identify pathogenicity of those uncertain significance variants.
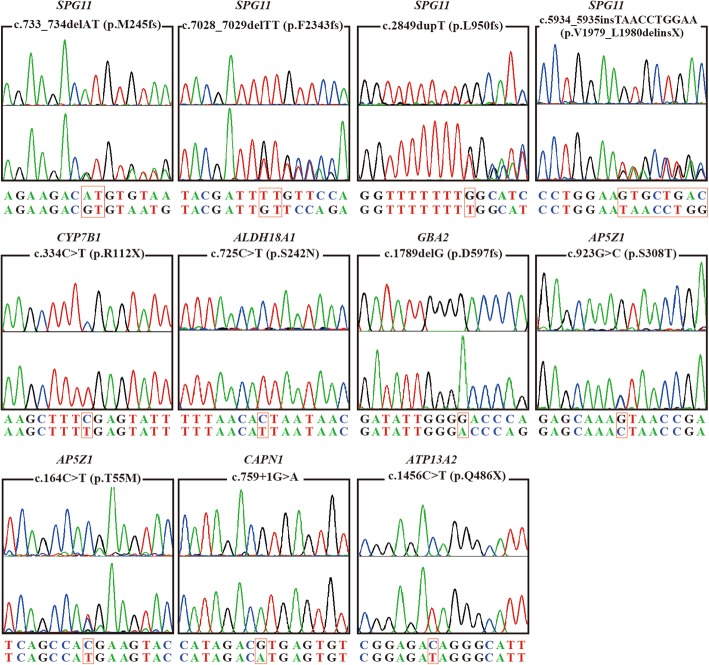
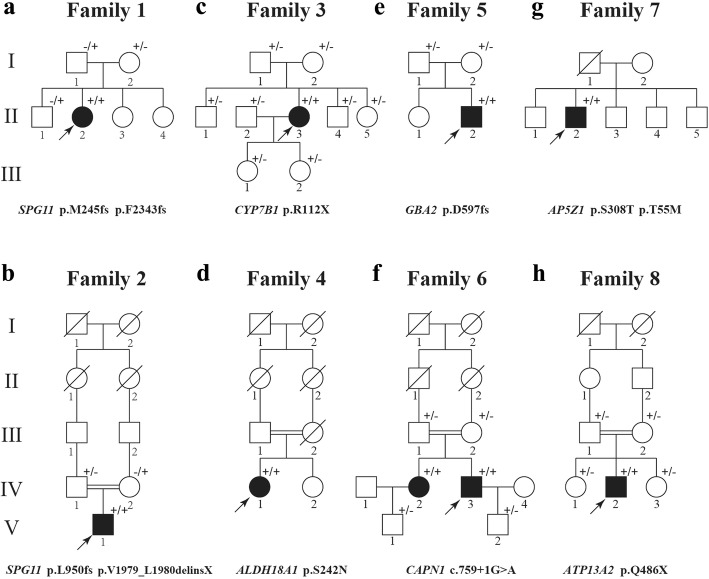
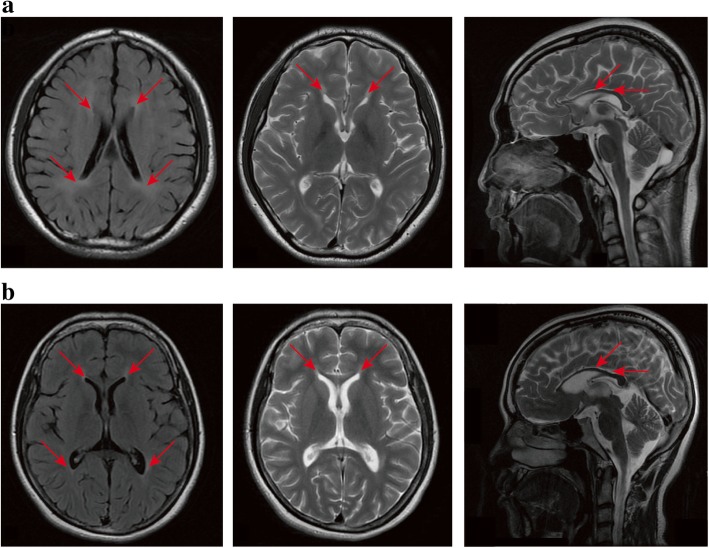
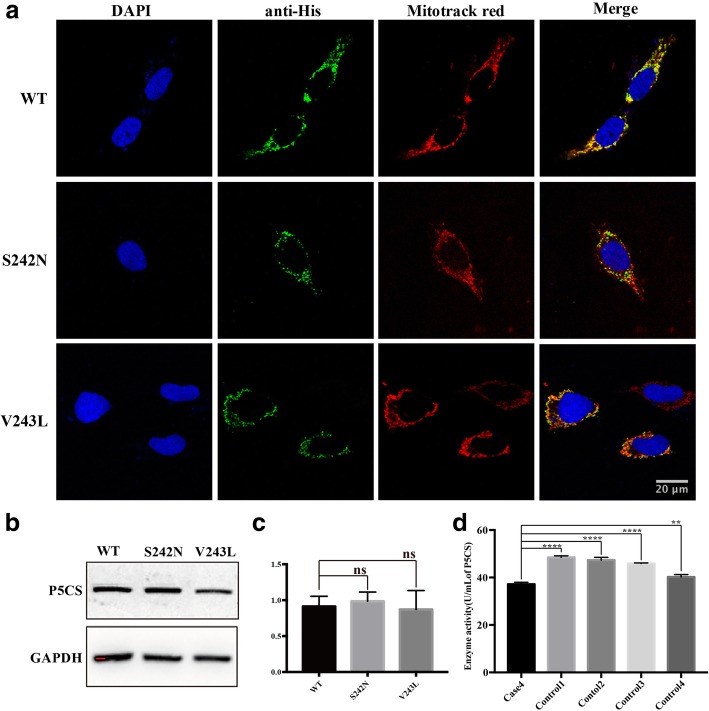
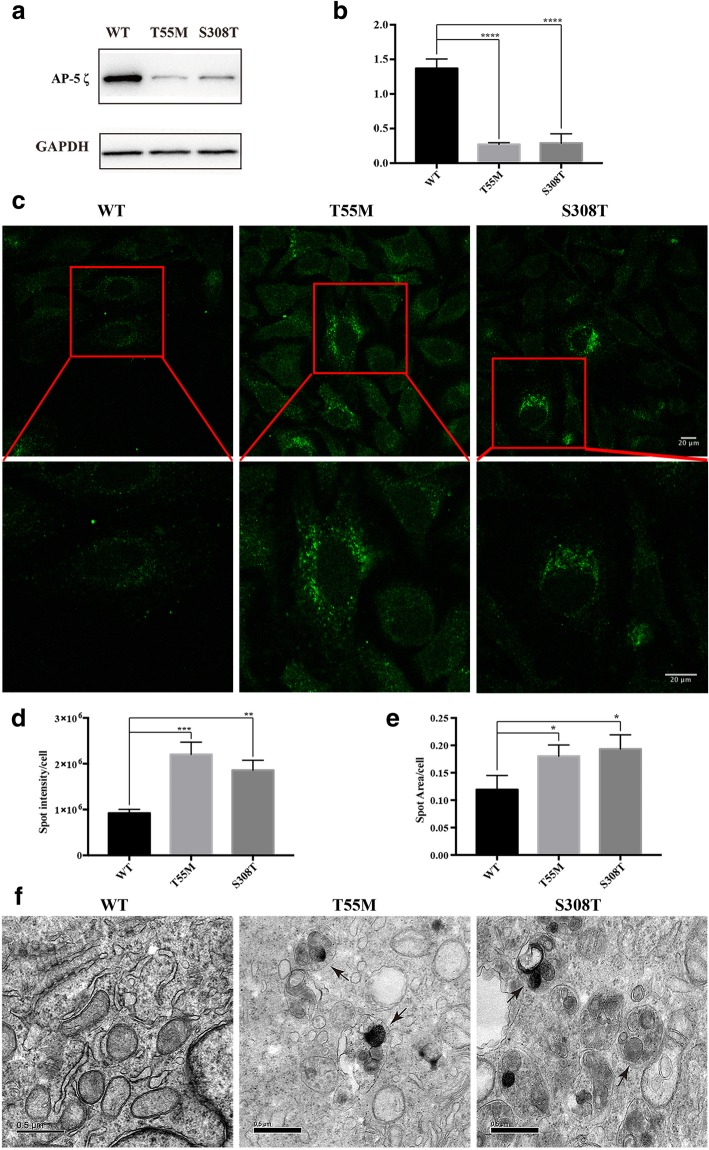
Results: We identified 11 mutations in HSP related genes including 7 novel mutations, including two (p.V1979_L1980delinsX, p.F2343 fs) in SPG11, two (p.T55 M, p.S308 T) in AP5Z1, one (p.S242 N) in ALDH18A1, one (p.D597fs) in GBA2, and one (p.Q486X) in ATP13A2 in 8 index patients and their family members. Mutations in ALDH18A1, AP5Z1, CAPN1 and ATP13A2 genes were firstly reported in the Chinese population. Furthermore, the clinical phenotypes of the patients carrying mutations were described in detail. The mutation (p.S242 N) in ALDH18A1 decreased enzyme activity of P5CS and mutations (p.T55 M, p.S308 T) in AP5Z1 induced lysosomal dysfunction.
Conclusion: Our results expanded the genetic spectrum and clinical profiles of AR-HSP patients and further demonstrated the efficiency and reliability of targeted NGS diagnosing suspected HSP patients.
Keywords: Autosomal recessive; Chinese; Genetic spectrum; Hereditary spastic paraplegia; Phenotype; Targeted next-generation sequencing.
Conflict of interest statement
Competing interestsThe authors declare that they have no competing interests.
Figures
Similar articles
-
Targeted next-generation sequencing improves diagnosis of hereditary spastic paraplegia in Chinese patients.J Mol Med (Berl). 2018 Jul;96(7):701-712. doi: 10.1007/s00109-018-1655-4. Epub 2018 Jun 11. J Mol Med (Berl). 2018. PMID: 29934652
-
Clinical features and genetic spectrum of Chinese patients with hereditary spastic paraplegia: A 14-year study.Front Genet. 2023 Feb 27;14:1085442. doi: 10.3389/fgene.2023.1085442. eCollection 2023. Front Genet. 2023. PMID: 36923789 Free PMC article.
-
Exome sequencing identifies novel compound heterozygous mutations in SPG11 that cause autosomal recessive hereditary spastic paraplegia.J Neurol Sci. 2013 Dec 15;335(1-2):112-7. doi: 10.1016/j.jns.2013.09.004. Epub 2013 Sep 10. J Neurol Sci. 2013. PMID: 24090761
-
Two novel homozygous mutations of CAPN1 in Chinese patients with hereditary spastic paraplegia and literatures review.Orphanet J Rare Dis. 2019 Apr 25;14(1):83. doi: 10.1186/s13023-019-1053-1. Orphanet J Rare Dis. 2019. PMID: 31023339 Free PMC article. Review.
-
More autosomal dominant SPG18 cases than recessive? The first AD-SPG18 pedigree in Chinese and literature review.Brain Behav. 2021 Dec;11(12):e32395. doi: 10.1002/brb3.2395. Epub 2021 Nov 3. Brain Behav. 2021. PMID: 34734492 Free PMC article. Review.
Cited by
-
Novel Compound Missense and Intronic Splicing Mutation in ALDH18A1 Causes Autosomal Recessive Spastic Paraplegia.Front Neurol. 2021 May 19;12:627531. doi: 10.3389/fneur.2021.627531. eCollection 2021. Front Neurol. 2021. PMID: 34093392 Free PMC article.
-
Two novel mutations in ALDH18A1 and SPG11 gene found by whole-exome sequencing in spastic paraplegia disease patients in Iran.Genomics Inform. 2022 Sep;20(3):e30. doi: 10.5808/gi.22030. Epub 2022 Sep 30. Genomics Inform. 2022. PMID: 36239107 Free PMC article.
-
Truncated mutants of beta-glucosidase 2 (GBA2) are localized in the mitochondrial matrix and cause mitochondrial fragmentation.PLoS One. 2020 Jun 3;15(6):e0233856. doi: 10.1371/journal.pone.0233856. eCollection 2020. PLoS One. 2020. PMID: 32492073 Free PMC article.
-
Hereditary spastic paraparesis type 46 (SPG46): new GBA2 variants in a large Italian case series and review of the literature.Neurogenetics. 2024 Apr;25(2):51-67. doi: 10.1007/s10048-024-00749-9. Epub 2024 Feb 9. Neurogenetics. 2024. PMID: 38334933 Free PMC article. Review.
-
Novel CAPN1 mutations extend the phenotypic heterogeneity in combined spastic paraplegia and ataxia.Ann Clin Transl Neurol. 2020 Oct;7(10):1862-1869. doi: 10.1002/acn3.51169. Epub 2020 Aug 29. Ann Clin Transl Neurol. 2020. PMID: 32860341 Free PMC article.
References
-
- Fink JK. Hereditary spastic paraplegia. Curr Neurol Neurosci Rep. 2006;6(1):65–76. - PubMed
-
- Harding AE. Classification of the hereditary ataxias and paraplegias. Lancet. 1983;1(8334):1151–1155. - PubMed
-
- Erichsen AK, Koht J, Stray-Pedersen A, Abdelnoor M, Tallaksen CM. Prevalence of hereditary ataxia and spastic paraplegia in Southeast Norway: a population-based study. Brain. 2009;132(Pt 6):1577–1588. - PubMed
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
