

Toxoplasma gondii induces prolonged host epidermal growth factor receptor signalling to prevent parasite elimination by autophagy: Perspectives for in vivo control of the parasite
- PMID: 31290228
- PMCID: PMC6771541
- DOI: 10.1111/cmi.13084
Toxoplasma gondii induces prolonged host epidermal growth factor receptor signalling to prevent parasite elimination by autophagy: Perspectives for in vivo control of the parasite
Abstract
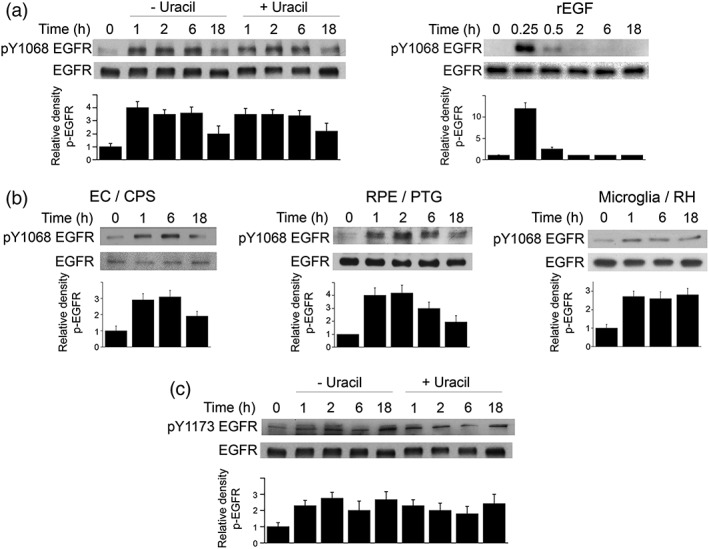
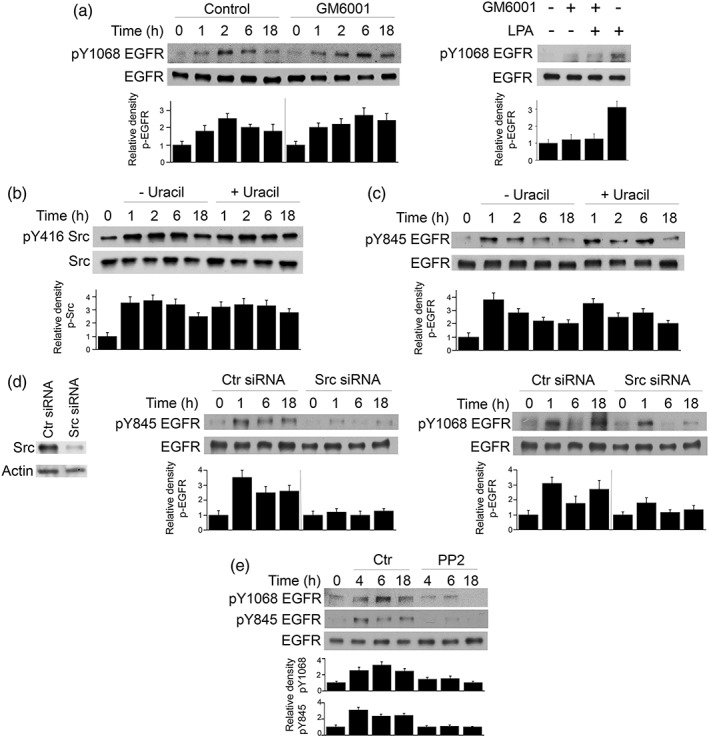
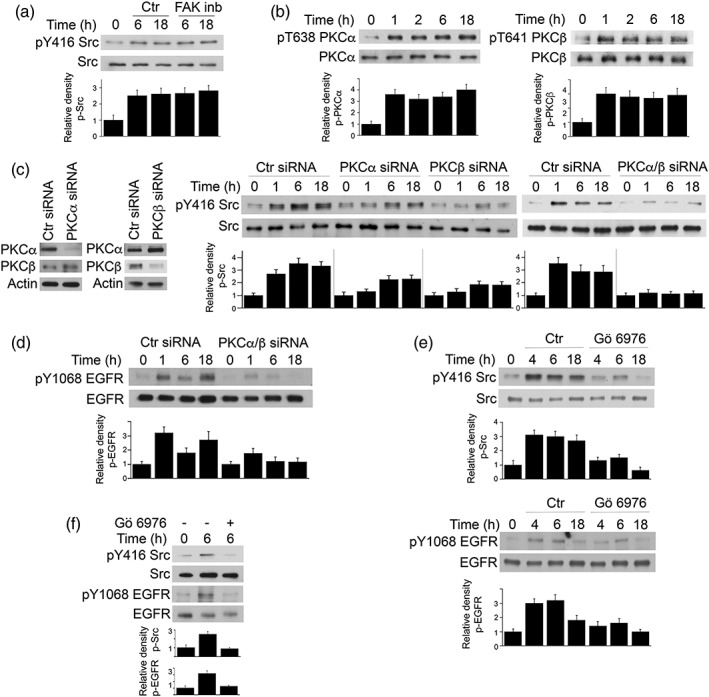
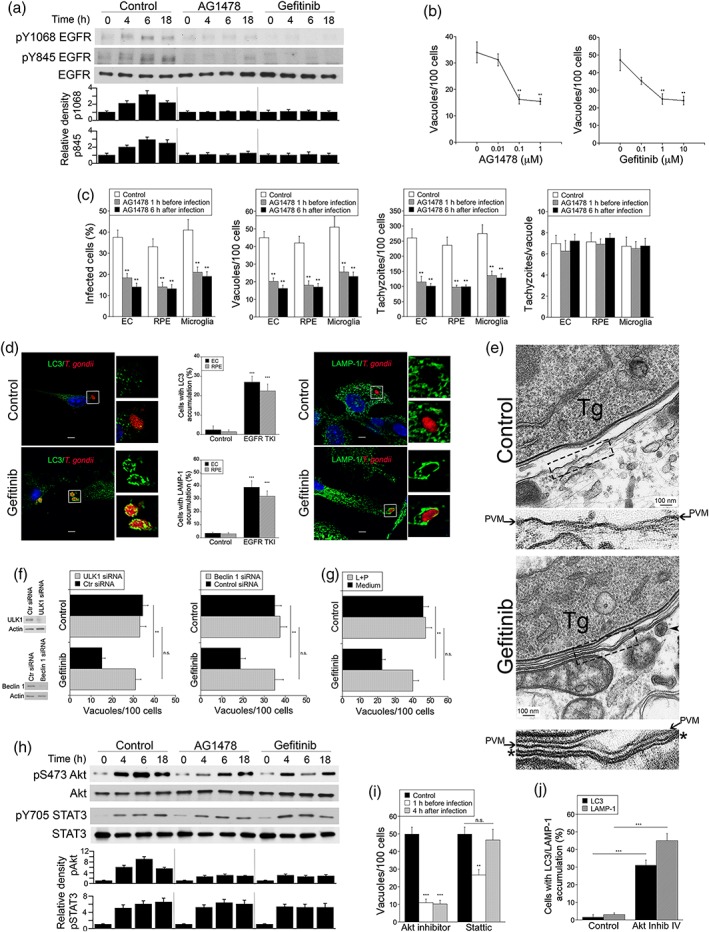
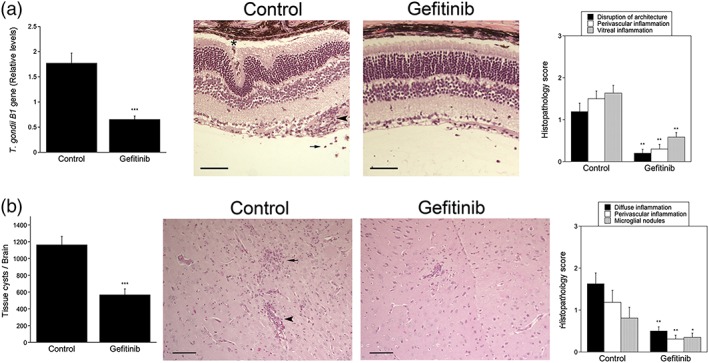
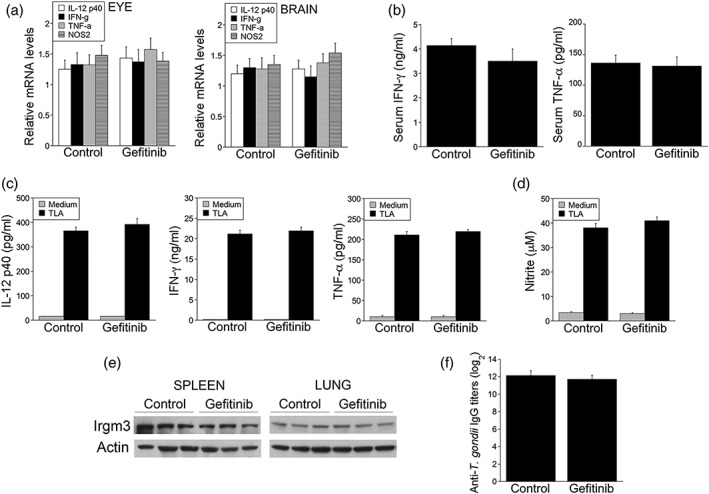
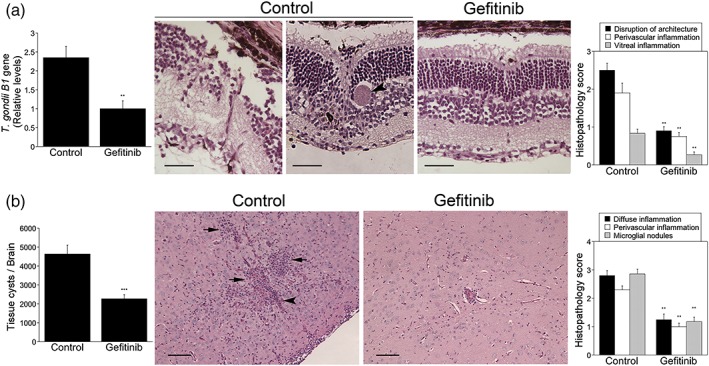
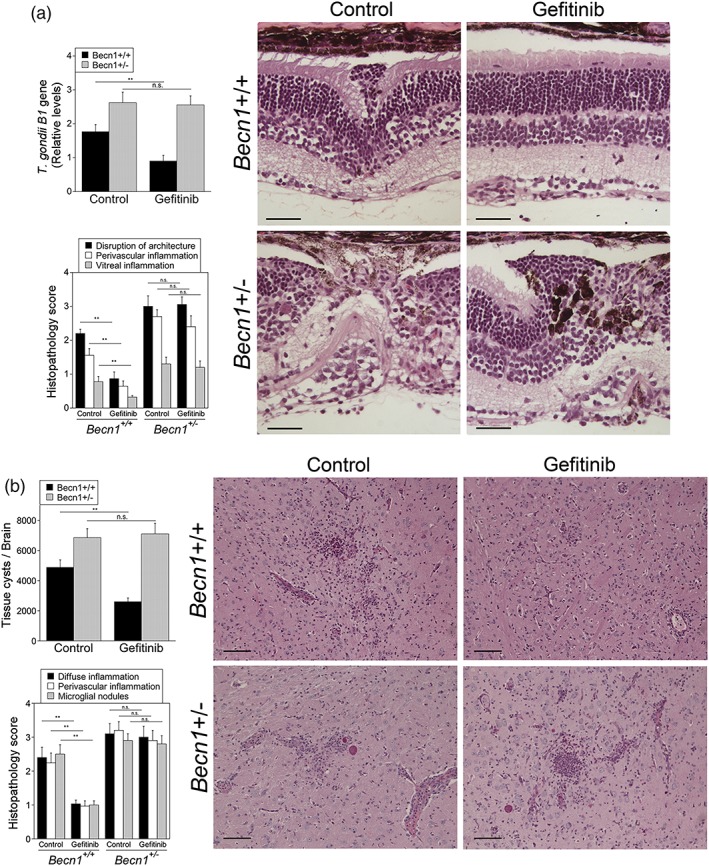
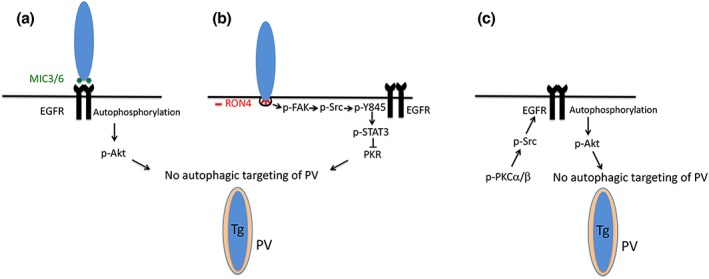
Toxoplasma gondii causes retinitis and encephalitis. Avoiding targeting by autophagosomes is key for its survival because T. gondii cannot withstand lysosomal degradation. During invasion of host cells, T. gondii triggers epidermal growth factor receptor (EGFR) signalling enabling the parasite to avoid initial autophagic targeting. However, autophagy is a constitutive process indicating that the parasite may also use a strategy operative beyond invasion to maintain blockade of autophagic targeting. Finding that such a strategy exists would be important because it could lead to inhibition of host cell signalling as a novel approach to kill the parasite in previously infected cells and treat toxoplasmosis. We report that T. gondii induced prolonged EGFR autophosphorylation. This effect was mediated by PKCα/PKCβ ➔ Src because T. gondii caused prolonged activation of these molecules and their knockdown or incubation with inhibitors of PKCα/PKCβ or Src after host cell invasion impaired sustained EGFR autophosphorylation. Addition of EGFR tyrosine kinase inhibitor (TKI) to previously infected cells led to parasite entrapment by LC3 and LAMP-1 and pathogen killing dependent on the autophagy proteins ULK1 and Beclin 1 as well as lysosomal enzymes. Administration of gefitinib (EGFR TKI) to mice with ocular and cerebral toxoplasmosis resulted in disease control that was dependent on Beclin 1. Thus, T. gondii promotes its survival through sustained EGFR signalling driven by PKCα/β ➔ Src, and inhibition of EGFR controls pre-established toxoplasmosis.
Keywords: infection; microbial-cell interaction; protozoa.
© 2019 The Authors Cellular Microbiology Published by John Wiley & Sons Ltd.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
References
-
- Andrade, R. M. , Wessendarp, M. , Gubbels, M. J. , Striepen, B. , & Subauste, C. S. (2006). CD40 induces macrophage anti‐Toxoplasma gondii activity by triggering autophagy‐dependent fusion of pathogen‐containing vacuoles and lysosomes. The Journal of Clinical Investigation, 116, 2366–2377. 10.1172/JCI28796 - DOI - PMC - PubMed
-
- Biscardi, J. S. , Maa, M. C. , Tice, D. A. , Cox, M. E. , Leu, T. H. , & Parsons, J. T. (1999). c‐Src‐mediated phosphorylation of the epidermal growth factor receptor on Tyr845 and Tyr1101 is associated with modulation of receptor function. The Journal of Biological Chemistry, 274, 8335–8343. 10.1074/jbc.274.12.8335 - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
