

Doxorubicin-induced cardiomyocyte death is mediated by unchecked mitochondrial fission and mitophagy
- PMID: 31291545
- PMCID: PMC6766652
- DOI: 10.1096/fj.201802663R
Doxorubicin-induced cardiomyocyte death is mediated by unchecked mitochondrial fission and mitophagy
Abstract
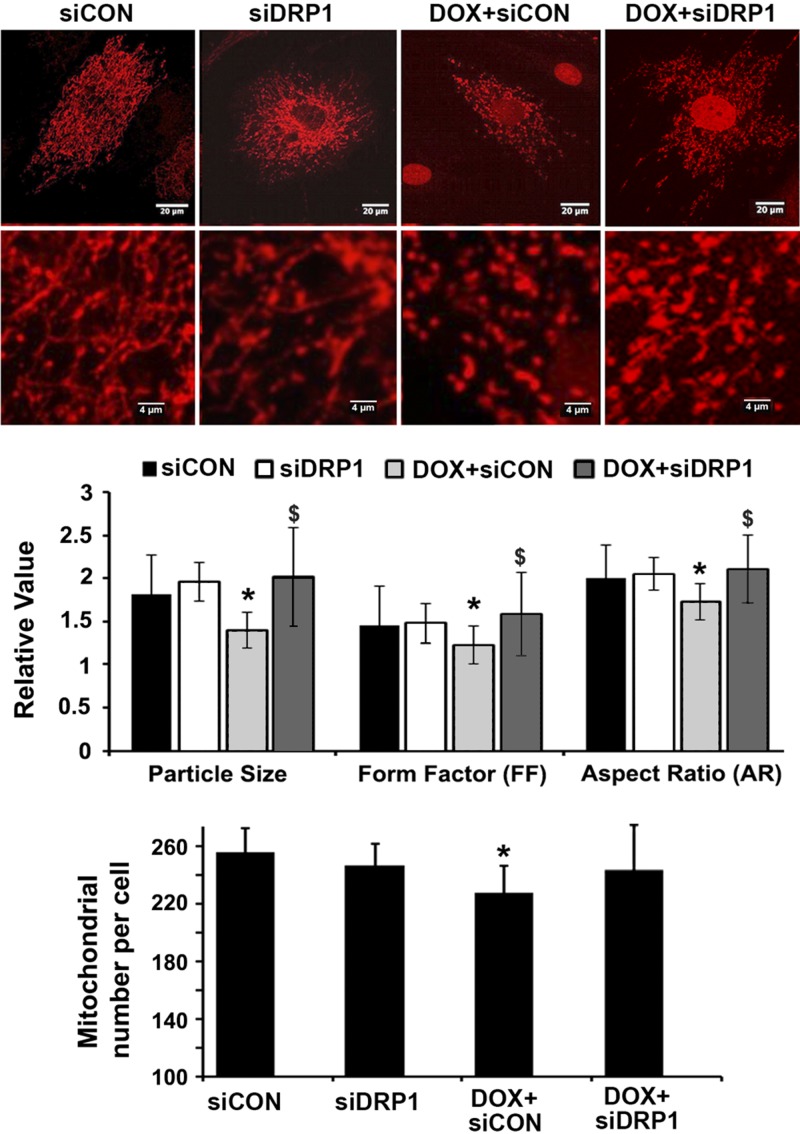
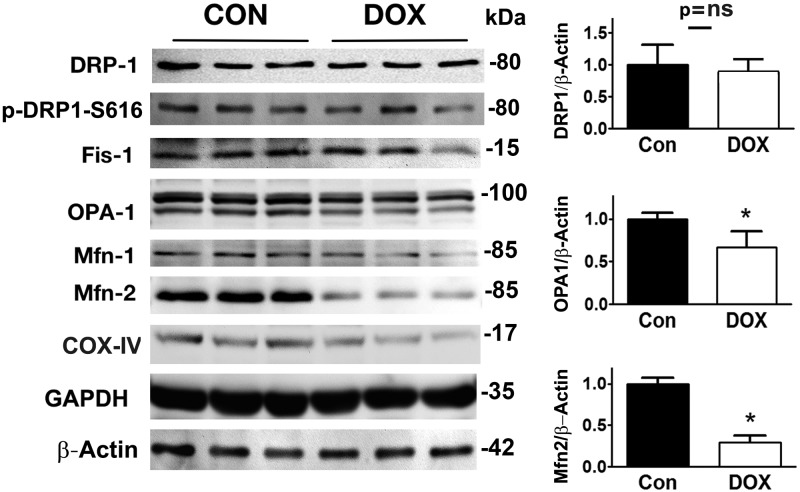
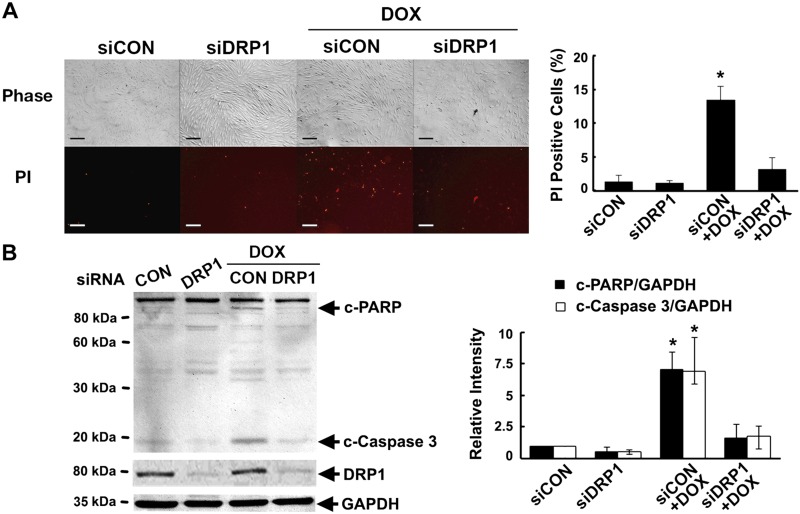
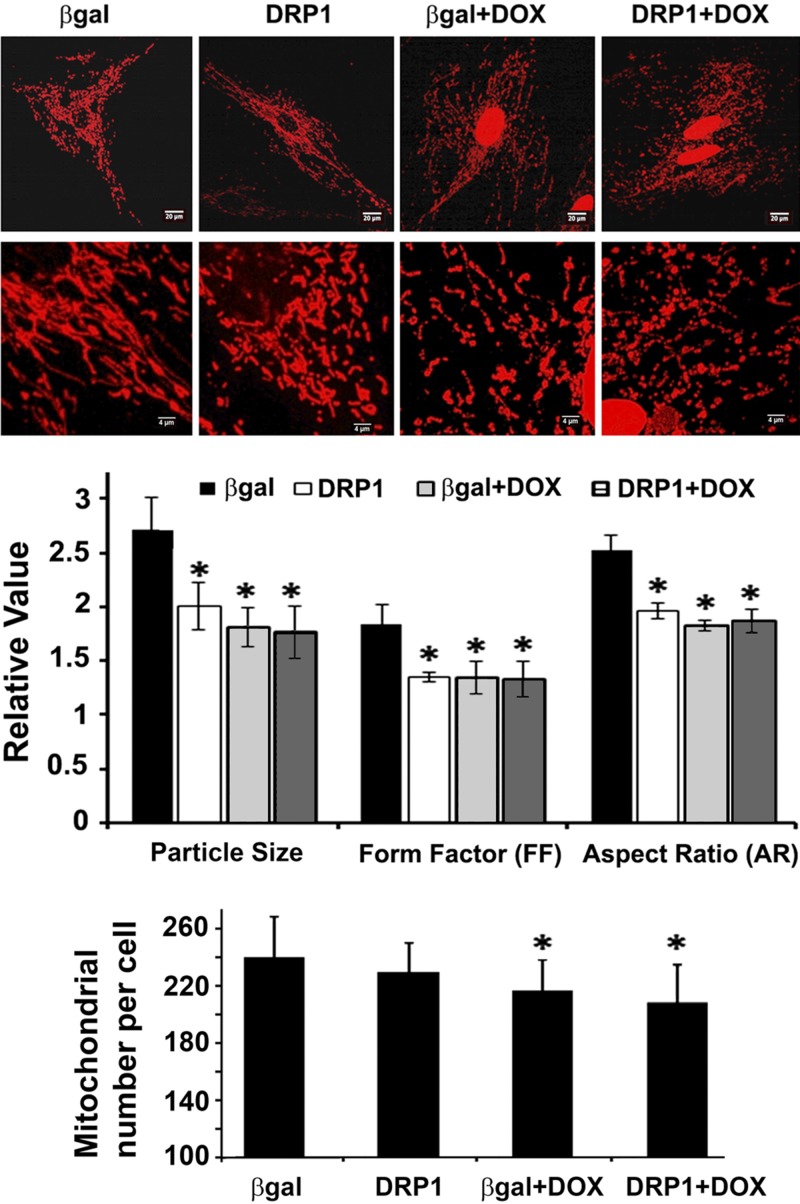
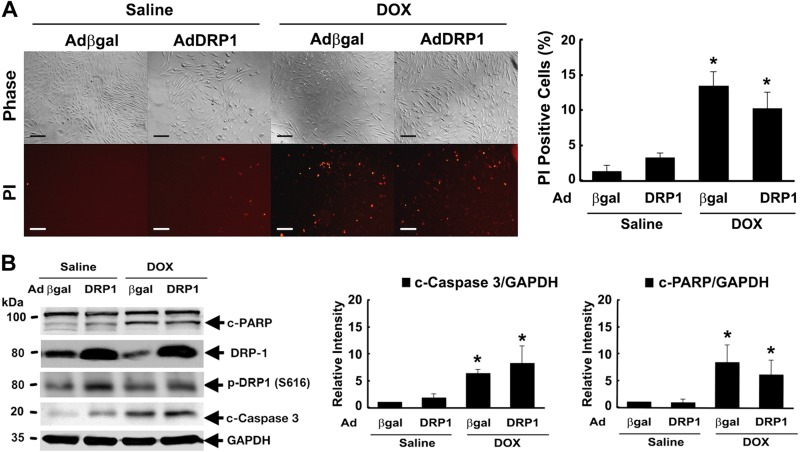
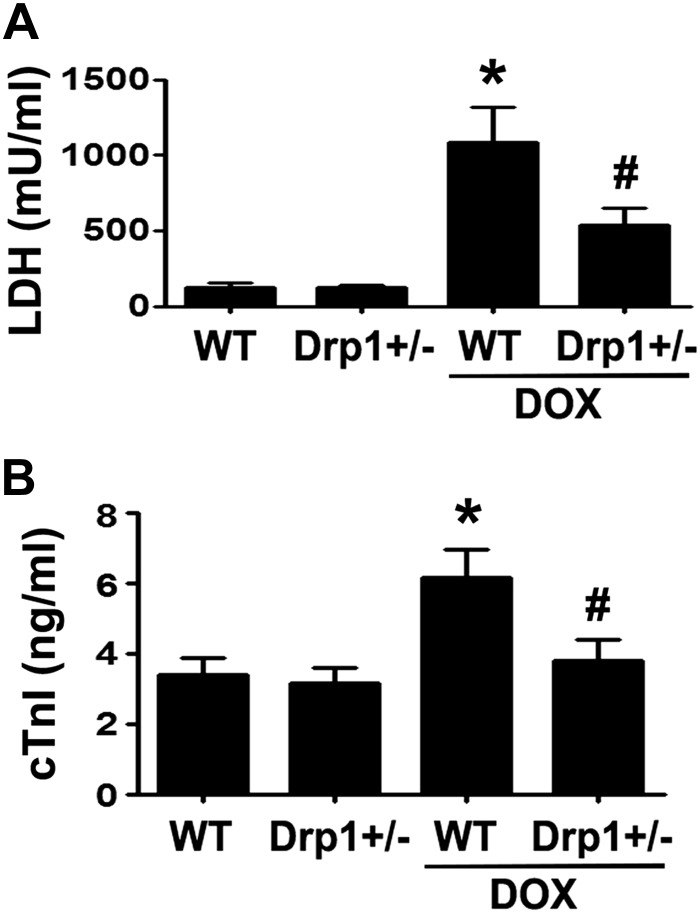
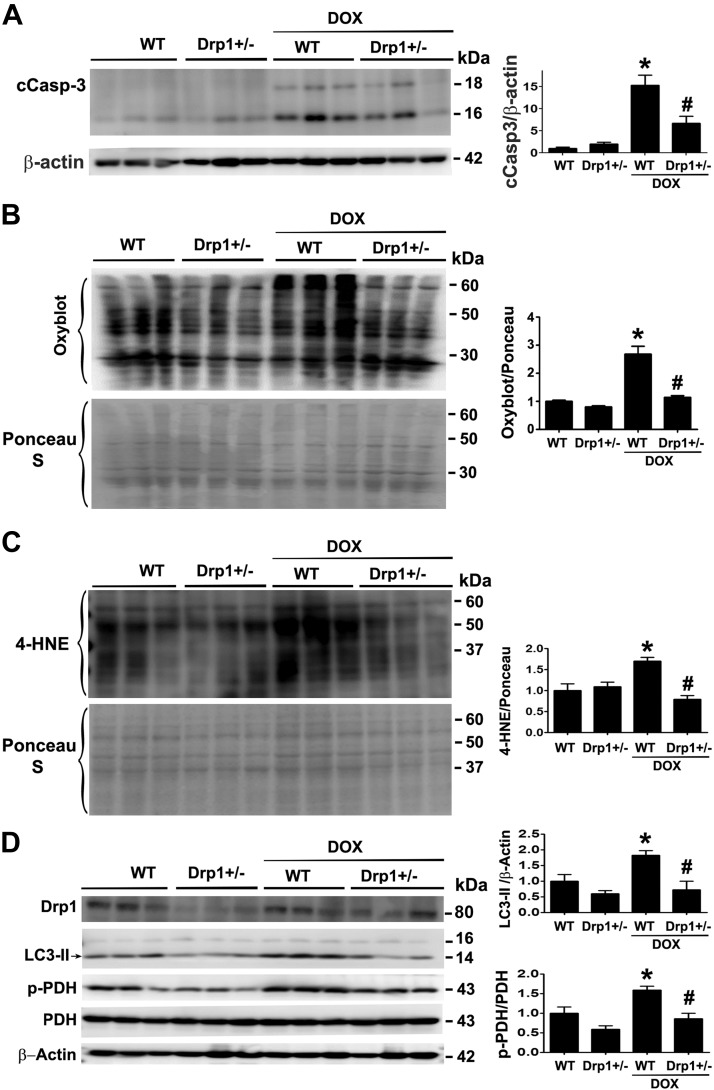
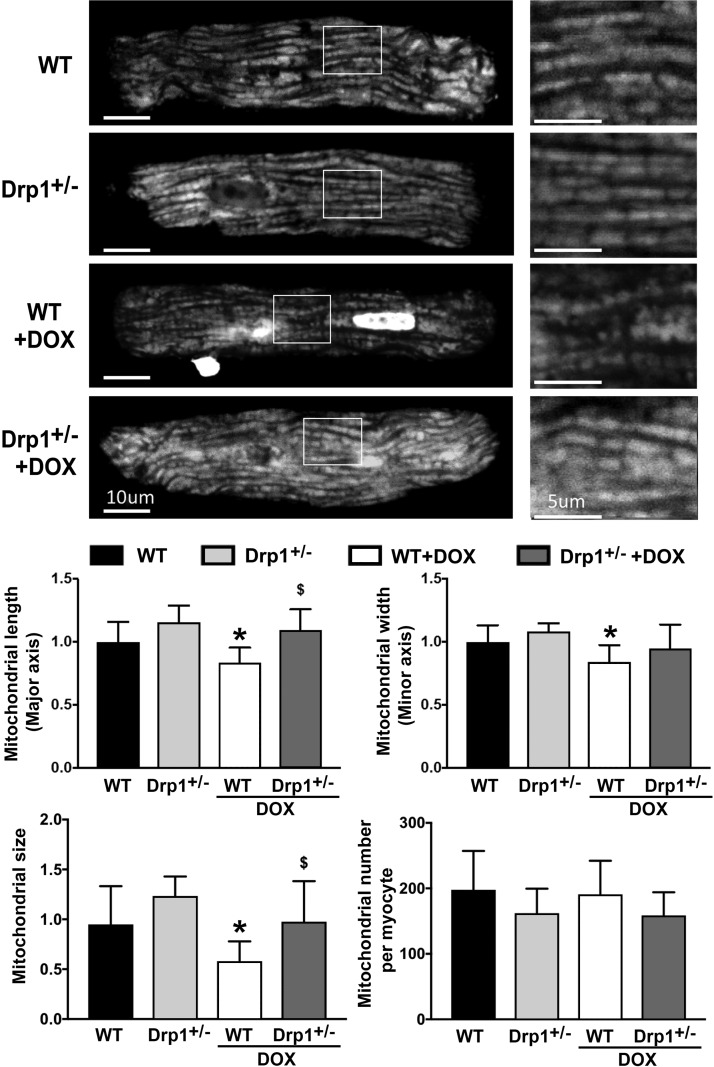
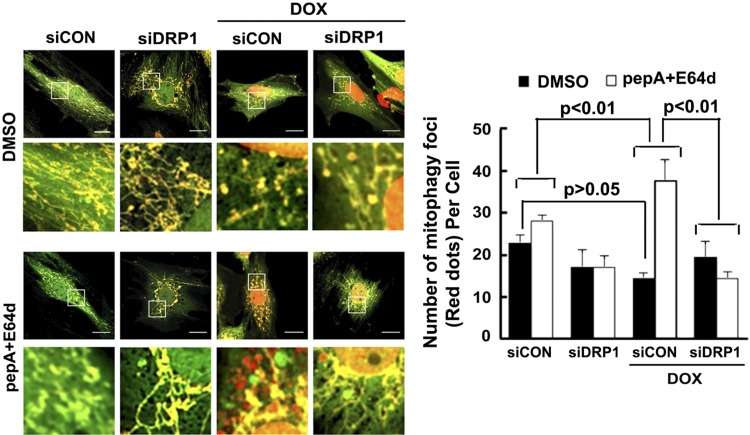
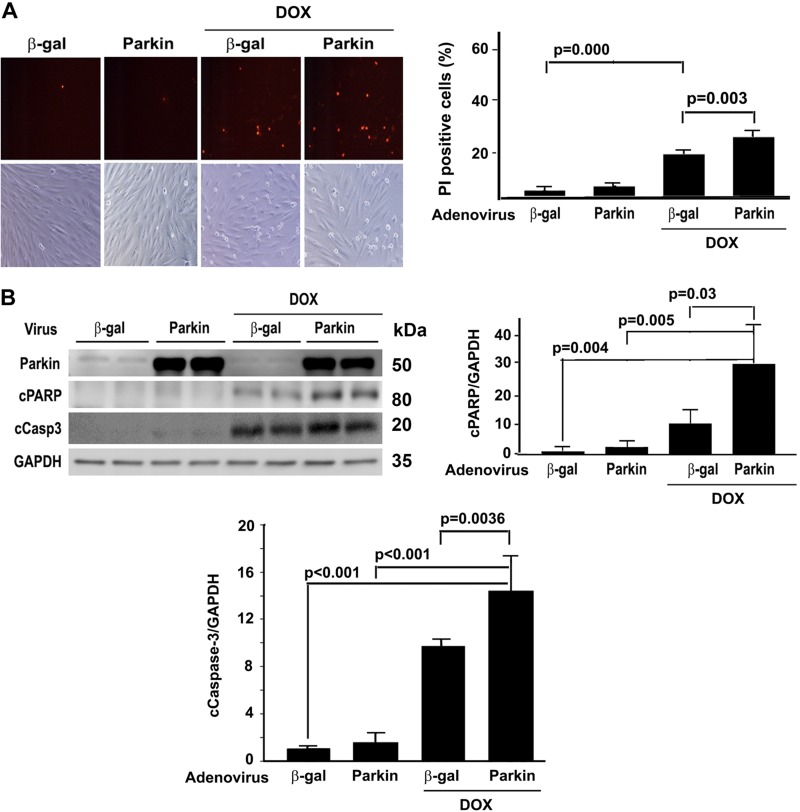
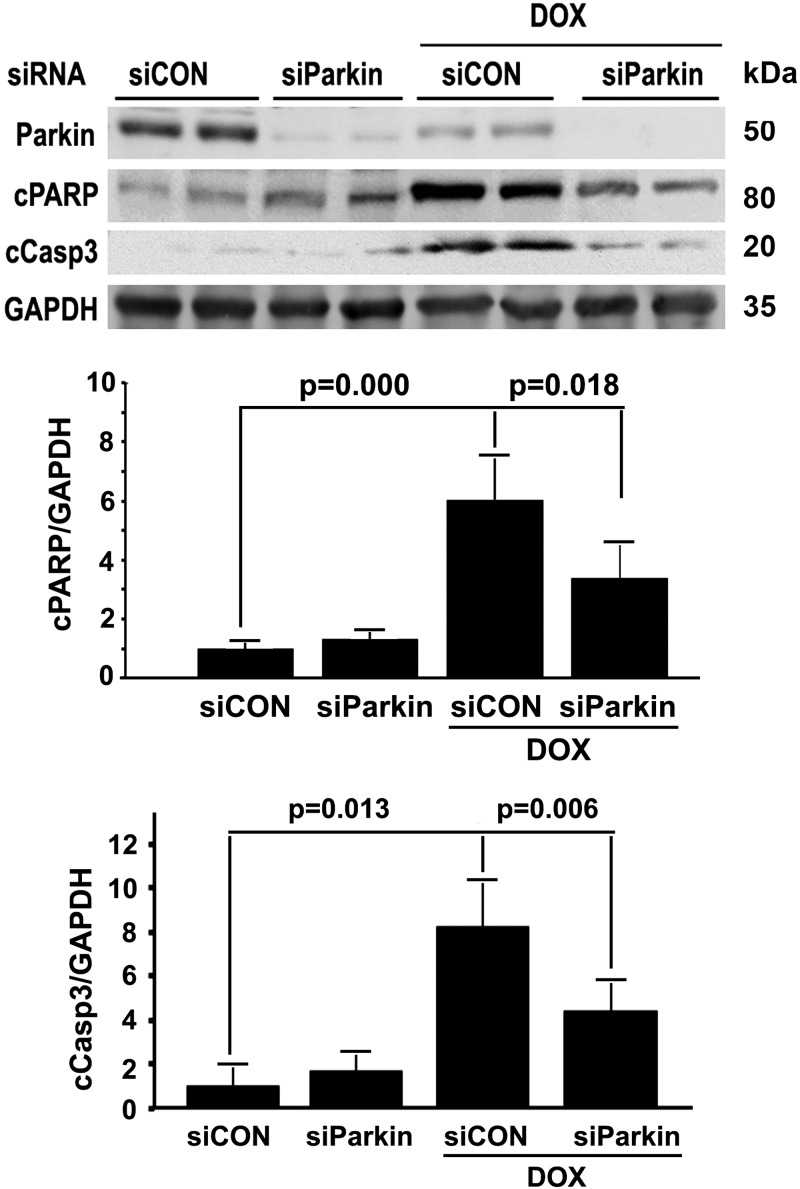
Doxorubicin (Dox) is a widely used antineoplastic agent that can cause heart failure. Dox cardiotoxicity is closely associated with mitochondrial damage. Mitochondrial fission and mitophagy are quality control mechanisms that normally help maintain a pool of healthy mitochondria. However, unchecked mitochondrial fission and mitophagy may compromise the viability of cardiomyocytes, predisposing them to cell death. Here, we tested this possibility by using Dox-treated H9c2 cardiac myoblast cells expressing either the mitochondria-targeted fluorescent protein MitoDsRed or the novel dual-fluorescent mitophagy reporter mt-Rosella. Dox induced mitochondrial fragmentation as shown by reduced form factor, aspect ratio, and mean mitochondrial size. This effect was abolished by short interference RNA-mediated knockdown of dynamin-related protein 1 (DRP1), a major regulator of fission. Importantly, DRP1 knockdown decreased cell death as indicated by the reduced number of propidium iodide-positive cells and the cleavage of caspase-3 and poly (ADP-ribose) polymerase. Moreover, DRP1-deficient mice were protected from Dox-induced cardiac damage, strongly supporting a role for DRP1-dependent mitochondrial fragmentation in Dox cardiotoxicity. In addition, Dox accelerated mitophagy flux, which was attenuated by DRP1 knockdown, as assessed by the mitophagy reporter mt-Rosella, suggesting the necessity of mitochondrial fragmentation in Dox-induced mitophagy. Knockdown of parkin, a positive regulator of mitophagy, dramatically diminished Dox-induced cell death, whereas overexpression of parkin had the opposite effect. Together, these results suggested that Dox cardiotoxicity was mediated, at least in part, by the increased mitochondrial fragmentation and accelerated mitochondrial degradation by the lysosome. Strategies that limit mitochondrial fission and mitophagy in the physiologic range may help reduce Dox cardiotoxicity.-Catanzaro, M. P., Weiner, A., Kaminaris, A., Li, C., Cai, F., Zhao, F., Kobayashi, S., Kobayashi, T., Huang, Y., Sesaki, H., Liang, Q. Doxorubicin-induced cardiomyocyte death is mediated by unchecked mitochondrial fission and mitophagy.
Keywords: anti-tumor agents; cardiotoxicity; heart failure; mitochondrial quality control.
Conflict of interest statement
Q.L. was supported by U.S. National Institutes of Health, National Heart, Lung, and Blood Institute Grant 1R15HL137130-01A1, and S.K. was supported by American Heart Association Grant 15SDG25080077. The authors declare no conflicts of interest.
Figures
References
-
- Swain S. M., Whaley F. S., Ewer M. S. (2003) Congestive heart failure in patients treated with doxorubicin: a retrospective analysis of three trials. Cancer 97, 2869–2879 - PubMed
-
- Minotti G., Menna P., Salvatorelli E., Cairo G., Gianni L. (2004) Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol. Rev. 56, 185–229 - PubMed
-
- Van der Pal H. J., van Dalen E. C., Hauptmann M., Kok W. E., Caron H. N., van den Bos C., Oldenburger F., Koning C. C., van Leeuwen F. E., Kremer L. C. (2010) Cardiac function in 5-year survivors of childhood cancer: a long-term follow-up study. Arch. Intern. Med. 170, 1247–1255 - PubMed
-
- Maeda M. (2008) Late effects of childhood cancer: life-threatening issues. J. Nippon Med. Sch. 75, 320–324 - PubMed
-
- Link G., Tirosh R., Pinson A., Hershko C. (1996) Role of iron in the potentiation of anthracycline cardiotoxicity: identification of heart cell mitochondria as a major site of iron-anthracycline interaction. J. Lab. Clin. Med. 127, 272–278 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
