

Tumour lineage shapes BRCA-mediated phenotypes
- PMID: 31292550
- PMCID: PMC7048239
- DOI: 10.1038/s41586-019-1382-1
Tumour lineage shapes BRCA-mediated phenotypes
Erratum in
-
Author Correction: Tumour lineage shapes BRCA-mediated phenotypes.Nature. 2020 Jan;577(7789):E1. doi: 10.1038/s41586-019-1839-2. Nature. 2020. PMID: 31822847
Abstract
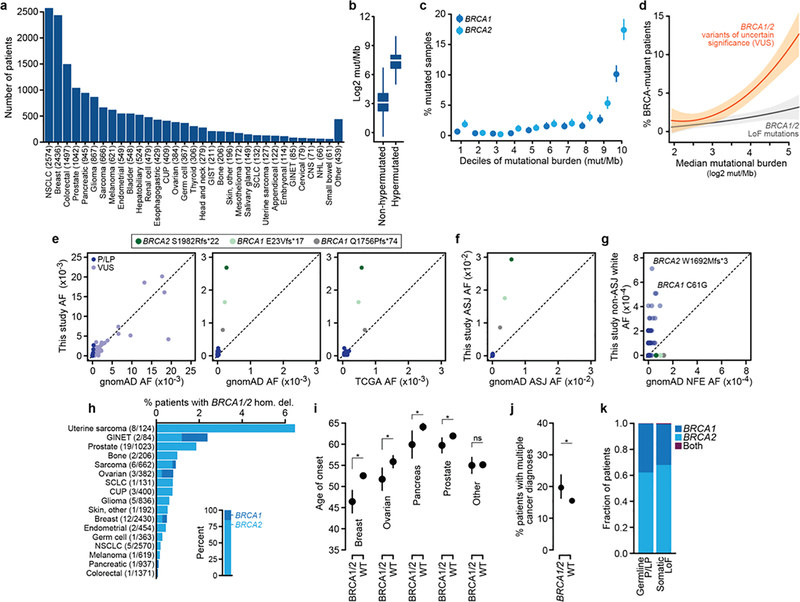
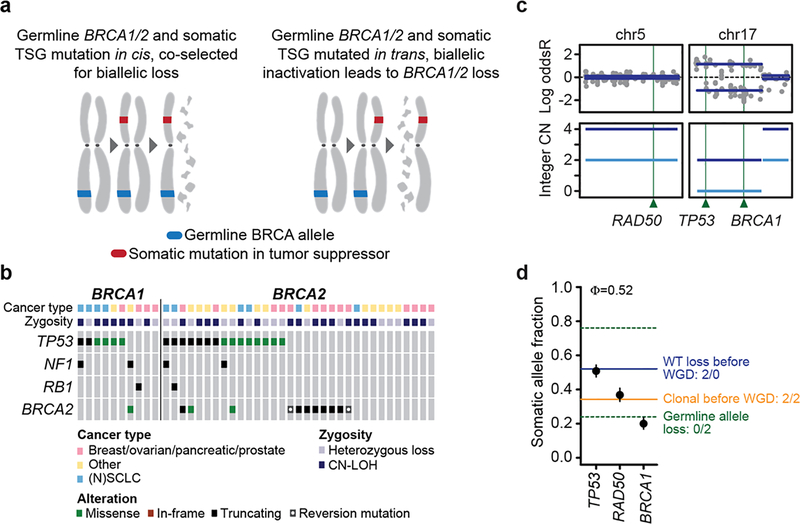
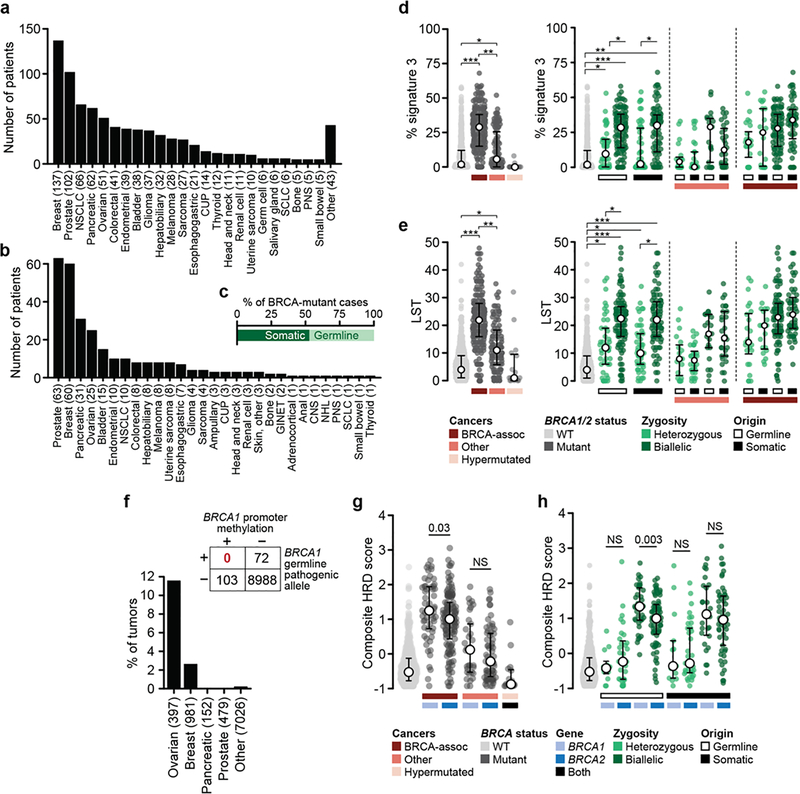
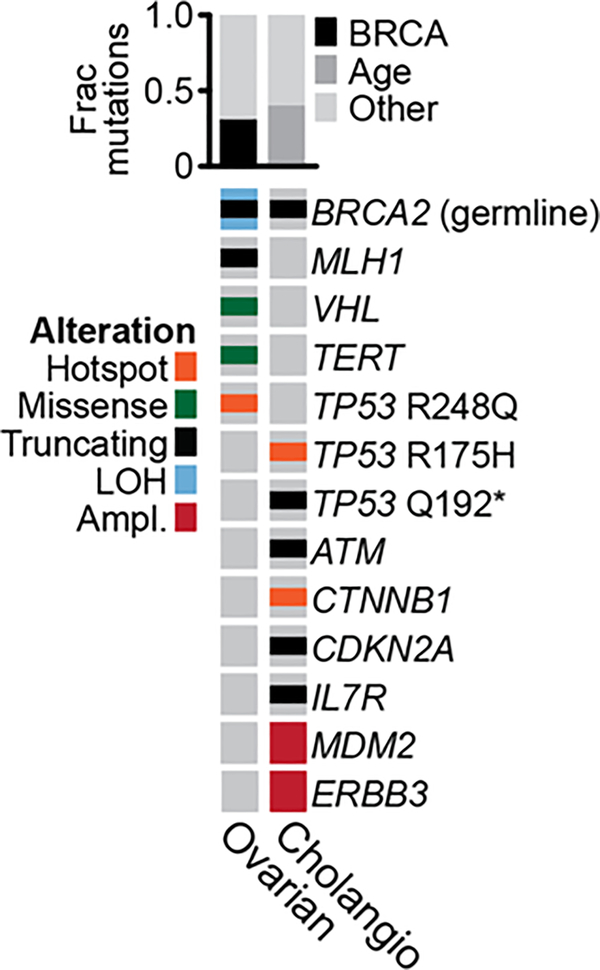
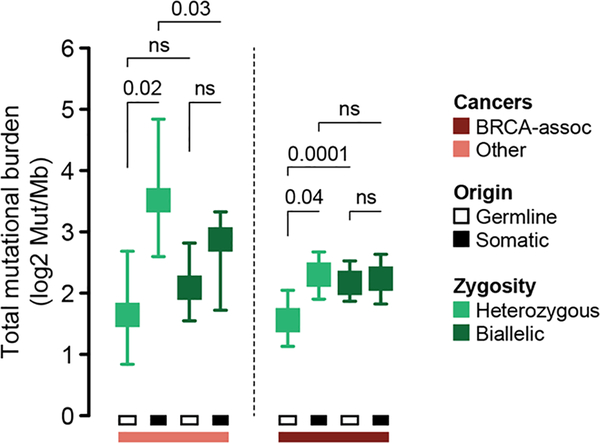
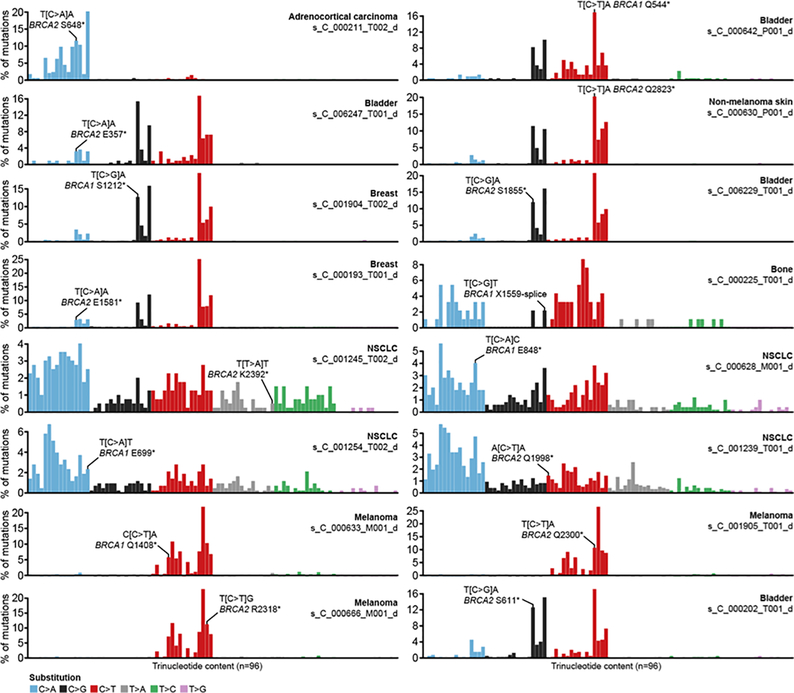
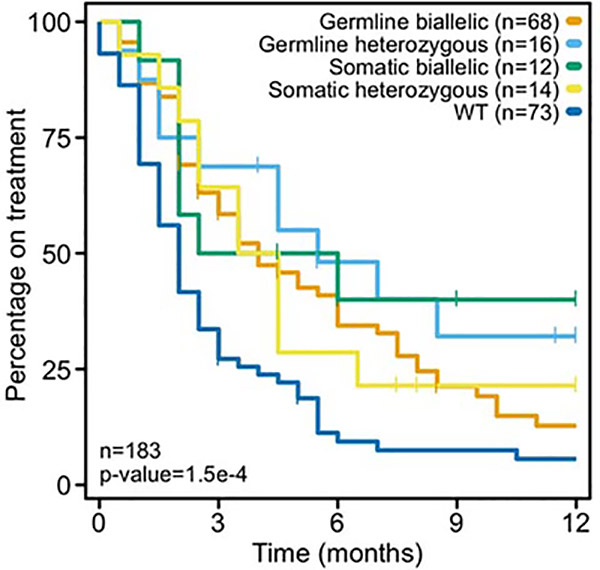
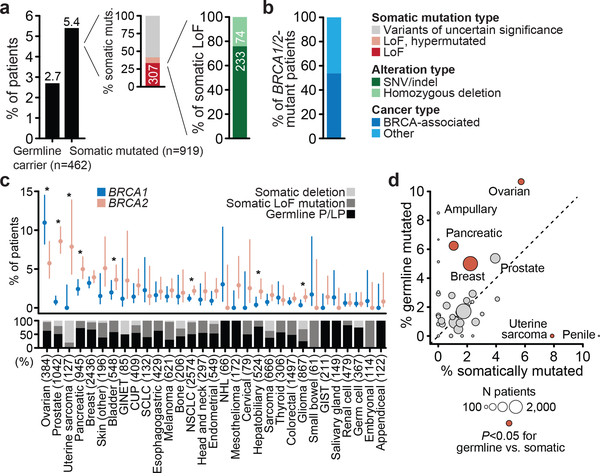
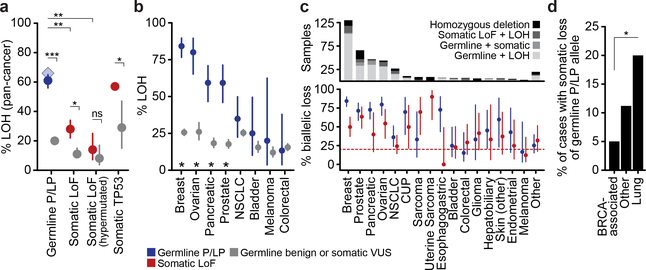
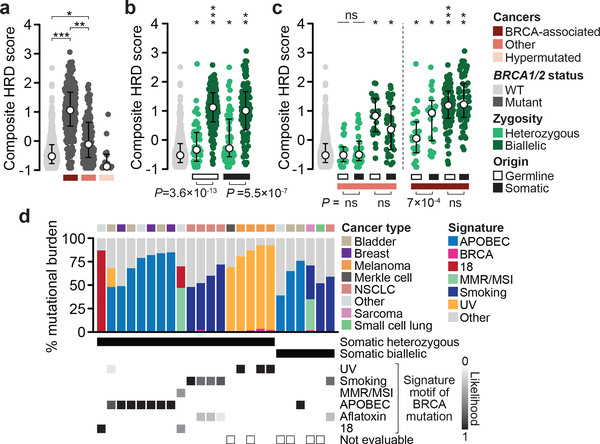
Mutations in BRCA1 and BRCA2 predispose individuals to certain cancers1-3, and disease-specific screening and preventative strategies have reduced cancer mortality in affected patients4,5. These classical tumour-suppressor genes have tumorigenic effects associated with somatic biallelic inactivation, although haploinsufficiency may also promote the formation and progression of tumours6,7. Moreover, BRCA1/2-mutant tumours are often deficient in the repair of double-stranded DNA breaks by homologous recombination8-13, and consequently exhibit increased therapeutic sensitivity to platinum-containing therapy and inhibitors of poly-(ADP-ribose)-polymerase (PARP)14,15. However, the phenotypic and therapeutic relevance of mutations in BRCA1 or BRCA2 remains poorly defined in most cancer types. Here we show that in the 2.7% and 1.8% of patients with advanced-stage cancer and germline pathogenic or somatic loss-of-function alterations in BRCA1/2, respectively, selective pressure for biallelic inactivation, zygosity-dependent phenotype penetrance, and sensitivity to PARP inhibition were observed only in tumour types associated with increased heritable cancer risk in BRCA1/2 carriers (BRCA-associated cancer types). Conversely, among patients with non-BRCA-associated cancer types, most carriers of these BRCA1/2 mutation types had evidence for tumour pathogenesis that was independent of mutant BRCA1/2. Overall, mutant BRCA is an indispensable founding event for some tumours, but in a considerable proportion of other cancers, it appears to be biologically neutral-a difference predominantly conditioned by tumour lineage-with implications for disease pathogenesis, screening, design of clinical trials and therapeutic decision-making.
Conflict of interest statement
Competing Interests
M.L.C. reports receiving travel/accommodation funding from Allergan, Sanofi-Aventis, Daiichi Sankyo. W.A. reports receiving honoraria from Caret, advisory board activities for Clovis Oncology, Janssen, and MORE Health, travel/accommodation expenses from Clovis Oncology and GlaxoSmithKline, and research funding from AstraZeneca, Zenith Epigenetics, Clovis Oncology, and GlaxoSmithKline. E.M.O. reports receiving consulting fees from BioLineRx, Targovax, Halozyme, Celgene, Cytomx, and Bayer and research funding support from Genentech, Roche, BMS, Halozyme, Celgene, MabVax Therapeutics, and ActaBiologica. D.M.H. reports receiving research funding from AstraZeneca, Puma Biotechnology, Loxo Oncology and personal fees from Atara Biotherapeutics, Chugai Pharma, Boehringer Ingelheim, AstraZeneca, Pfizer, Bayer, Debiophram Group, and Genetech. M.F.B. reports receiving research funding from Illumina and advisory board activities for Roche. D.B.S. reports advisory board activities for Loxo Oncology, Pfizer, Illumina, Lilly Oncology, Vivideon, and Intezyne. All stated activities were outside of the work described herein. No other disclosures were noted.
Figures


Comment in
-
Why BRCA mutations are not tumour-agnostic biomarkers for PARP inhibitor therapy.Nat Rev Clin Oncol. 2019 Dec;16(12):725-726. doi: 10.1038/s41571-019-0285-2. Nat Rev Clin Oncol. 2019. PMID: 31582817 No abstract available.
-
Melanoma predisposition-A limited role for germline BRCA1 and BRCA2 variants.Pigment Cell Melanoma Res. 2020 Jan;33(1):6-7. doi: 10.1111/pcmr.12833. Epub 2019 Oct 28. Pigment Cell Melanoma Res. 2020. PMID: 31610093 Free PMC article. No abstract available.
-
Etiologic Index - A Case-Only Measure of BRCA1/2-Associated Cancer Risk.N Engl J Med. 2020 Jul 16;383(3):286-288. doi: 10.1056/NEJMc1913988. N Engl J Med. 2020. PMID: 32668122 Free PMC article. No abstract available.
-
Heterozygous PALB2 Mutation in a Boy with Acute Lymphoblastic Leukemia and Subsequent Metastatic Ewing Sarcoma.Klin Padiatr. 2021 May;233(3):141-144. doi: 10.1055/a-1404-3243. Epub 2021 Mar 26. Klin Padiatr. 2021. PMID: 33772500 English. No abstract available.
References
-
- Wooster R et al. Identification of the breast cancer susceptibility gene BRCA2. Nature 378, 789–792 (1995). - PubMed
-
- Miki Y et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 266, 66–71 (1994). - PubMed
-
- Kuchenbaecker KB et al. Risks of breast, ovarian, and contralateral breast cancer for BRCA1 and BRCA2 mutation carriers. JAMA 317, 2402–2416 (2017). - PubMed
-
- Paluch-Shimon S et al. Prevention and screening in BRCA mutation carriers and other breast/ovarian hereditary cancer syndromes: ESMO Clinical Practice Guidelines for cancer prevention and screening. Ann. Oncol 27, v103–v110 (2016). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
