

The Landscape of Persistent Viral Genomes in ART-Treated SIV, SHIV, and HIV-2 Infections
- PMID: 31295427
- PMCID: PMC6724192
- DOI: 10.1016/j.chom.2019.06.005
The Landscape of Persistent Viral Genomes in ART-Treated SIV, SHIV, and HIV-2 Infections
Abstract
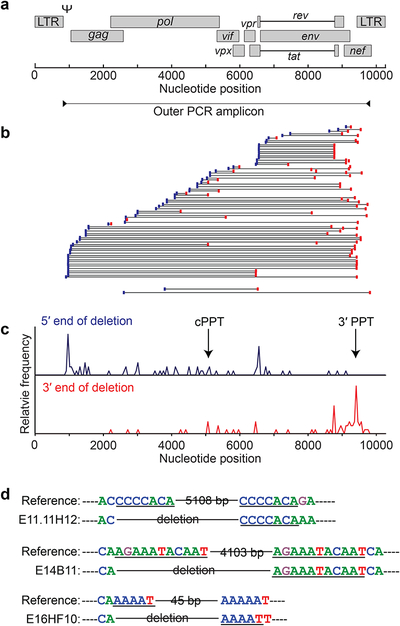
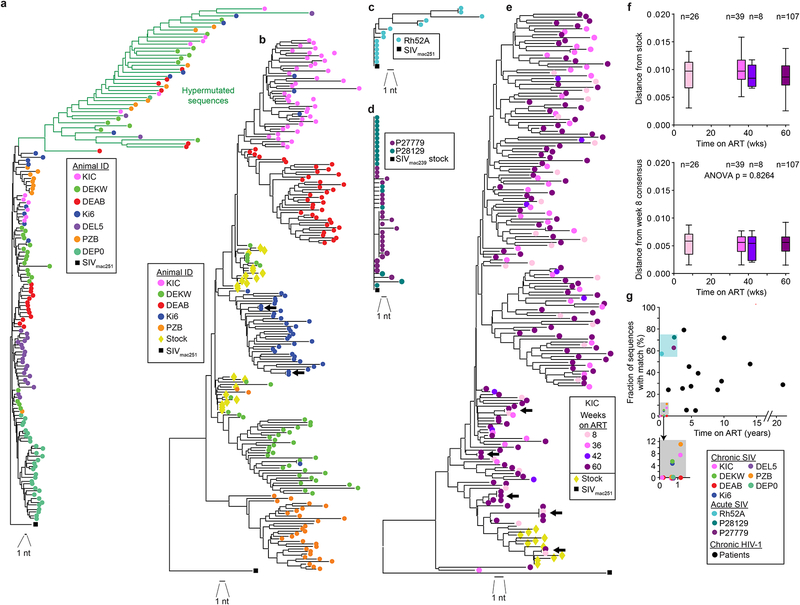
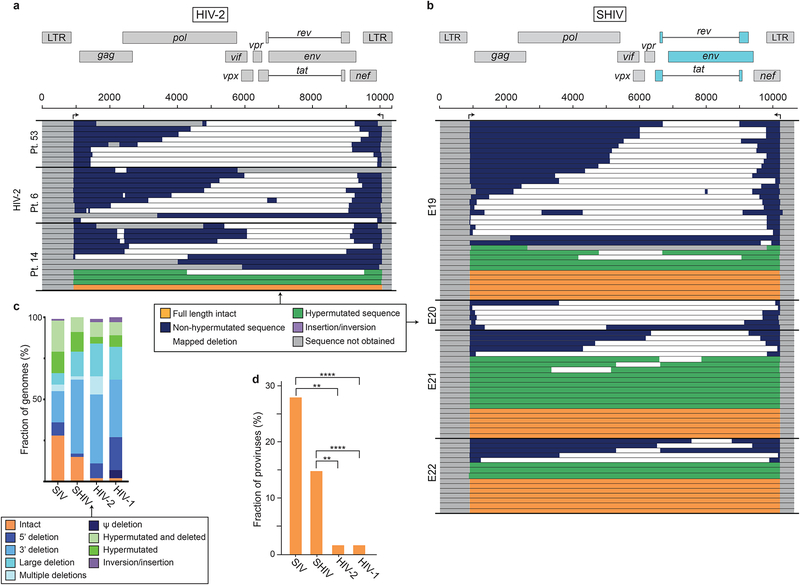
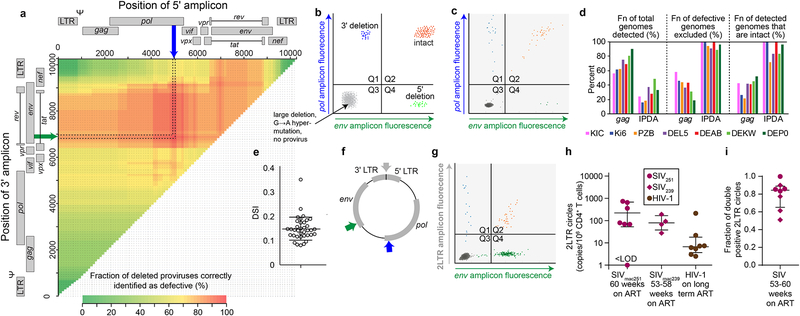
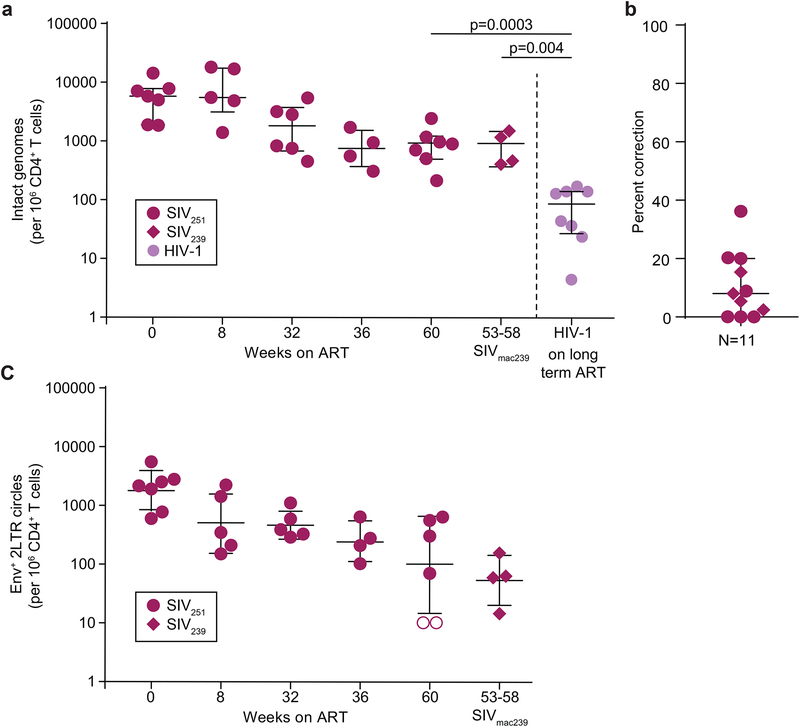
Evaluation of HIV cure strategies is complicated by defective proviruses that persist in ART-treated patients but are irrelevant to cure. Non-human primates (NHP) are essential for testing cure strategies. However, the persisting proviral landscape in ART-treated NHPs is uncharacterized. Here, we describe viral genomes persisting in ART-treated, simian immunodeficiency virus (SIV)-infected NHPs, simian-human immunodeficiency virus (SHIV)-infected NHPs, and humans infected with HIV-2, an SIV-related virus. The landscapes of persisting SIV, SHIV, and HIV-2 genomes are also dominated by defective sequences. However, there was a significantly higher fraction of intact SIV proviral genomes compared to ART-treated HIV-1 or HIV-2 infected humans. Compared to humans with HIV-1, SIV-infected NHPs had more hypermutated genomes, a relative paucity of clonal SIV sequences, and a lower frequency of deleted genomes. Finally, we report an assay for measuring intact SIV genomes which may have value in cure research.
Keywords: HIV-2; SHIV; SIV; clonal expansion; defective provirus; latent reservoir.
Copyright © 2019 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests
Aspects of HIV-1 IPDA are subject of a patent application PCT/US16/28822 filed by Johns Hopkins University. K.M.B. and R.F.S. are inventors on this application. Accelevir Diagnostics holds an exclusive license for this patent application. G.M.L. is an employee of and shareholder in Accelevir Diagnostics. R.F.S. holds no equity interest in Accelevir Diagnostics. R.F.S is a consultant on cure-related HIV research for Merck and Abbvie.
Figures


Comment in
-
For Viral Reservoir Studies, Timing Matters.Trends Microbiol. 2019 Oct;27(10):809-810. doi: 10.1016/j.tim.2019.08.003. Epub 2019 Aug 17. Trends Microbiol. 2019. PMID: 31431318
Similar articles
-
Biphasic decay of intact SHIV genomes following initiation of antiretroviral therapy complicates analysis of interventions targeting the reservoir.Proc Natl Acad Sci U S A. 2023 Oct 24;120(43):e2313209120. doi: 10.1073/pnas.2313209120. Epub 2023 Oct 16. Proc Natl Acad Sci U S A. 2023. PMID: 37844236 Free PMC article.
-
Simian-Human Immunodeficiency Virus SHIV.C.CH505 Persistence in ART-Suppressed Infant Macaques Is Characterized by Elevated SHIV RNA in the Gut and a High Abundance of Intact SHIV DNA in Naive CD4+ T Cells.J Virol. 2020 Dec 22;95(2):e01669-20. doi: 10.1128/JVI.01669-20. Print 2020 Dec 22. J Virol. 2020. PMID: 33087463 Free PMC article.
-
Evaluating the Intactness of Persistent Viral Genomes in Simian Immunodeficiency Virus-Infected Rhesus Macaques after Initiating Antiretroviral Therapy within One Year of Infection.J Virol. 2019 Dec 12;94(1):e01308-19. doi: 10.1128/JVI.01308-19. Print 2019 Dec 12. J Virol. 2019. PMID: 31597776 Free PMC article.
-
Quantitative and Qualitative Distinctions between HIV-1 and SIV Reservoirs: Implications for HIV-1 Cure-Related Studies.Viruses. 2024 Mar 27;16(4):514. doi: 10.3390/v16040514. Viruses. 2024. PMID: 38675857 Free PMC article. Review.
-
Brain macrophages harbor latent, infectious simian immunodeficiency virus.AIDS. 2019 Dec 1;33 Suppl 2(Suppl 2):S181-S188. doi: 10.1097/QAD.0000000000002269. AIDS. 2019. PMID: 31789817 Free PMC article. Review.
Cited by
-
Pharmacological Validation of Long-Term Treatment with Antiretroviral Drugs in a Model of SIV-Infected Non-Human Primates.Pharmaceutics. 2022 Oct 25;14(11):2282. doi: 10.3390/pharmaceutics14112282. Pharmaceutics. 2022. PMID: 36365101 Free PMC article.
-
Intact proviral DNA assay analysis of large cohorts of people with HIV provides a benchmark for the frequency and composition of persistent proviral DNA.Proc Natl Acad Sci U S A. 2020 Aug 4;117(31):18692-18700. doi: 10.1073/pnas.2006816117. Epub 2020 Jul 20. Proc Natl Acad Sci U S A. 2020. PMID: 32690683 Free PMC article.
-
Measuring the Haystack's Needles.J Infect Dis. 2021 Feb 3;223(2):184-186. doi: 10.1093/infdis/jiaa533. J Infect Dis. 2021. PMID: 32823275 Free PMC article. No abstract available.
-
CD8+ cells and small viral reservoirs facilitate post-ART control of SIV replication in M3+ Mauritian cynomolgus macaques initiated on ART two weeks post-infection.PLoS Pathog. 2023 Sep 25;19(9):e1011676. doi: 10.1371/journal.ppat.1011676. eCollection 2023 Sep. PLoS Pathog. 2023. PMID: 37747933 Free PMC article.
-
BCL-2 Inhibition via Venetoclax at ART Initiation Induces Long-Term Reduction of the Intact SIV Reservoir.Res Sq [Preprint]. 2025 Jul 14:rs.3.rs-7060088. doi: 10.21203/rs.3.rs-7060088/v1. Res Sq. 2025. PMID: 40709247 Free PMC article. Preprint.
References
-
- Ambrose Z, Palmer S, Boltz VF, Kearney M, Larsen K, Polacino P, Flanary L, Oswald K, Piatak M Jr, Smedley J, et al. (2007). Suppression of viremia and evolution of human immunodeficiency virus type 1 drug resistance in a macaque model for antiretroviral therapy. J. Virol 81, 12145–12155. - PMC - PubMed
-
- Bailey JR, Sedaghat AR, Kieffer T, Brennan T, Lee PK, Wind-Rotolo M, Haggerty CM, Kamireddi AR, Liu Y, Lee J, et al. (2006). Residual human immunodeficiency virus type 1 viremia in some patients on antiretroviral therapy is dominated by a small number of invariant clones rarely found in circulating CD4+ T cells. J. Virol 80, 6441–6457. - PMC - PubMed
-
- Balzarini J, Weeger M, Camarasa MJ, De Clercq E, and Liberia K (1995). Sensitivity/resistance profile of a simian immunodeficiency virus containing the reverse transcriptase gene of human immunodeficiency virus type 1 (HIV-1) toward the HIV-1-specific non-nucleoside reverse transcriptase inhibitors. Biochem. Biophys. Res. Commun 211, 850–856. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- UM1 AI126620/AI/NIAID NIH HHS/United States
- R01 AI120765/AI/NIAID NIH HHS/United States
- T32 AI007291/AI/NIAID NIH HHS/United States
- P01 AI131306/AI/NIAID NIH HHS/United States
- UL1 TR001863/TR/NCATS NIH HHS/United States
- UM1 AI126603/AI/NIAID NIH HHS/United States
- P01 MH070306/MH/NIMH NIH HHS/United States
- U42 OD010426/OD/NIH HHS/United States
- R01 AI127142/AI/NIAID NIH HHS/United States
- HHMI/Howard Hughes Medical Institute/United States
- R01 AI051178/AI/NIAID NIH HHS/United States
- R01 NS076357/NS/NINDS NIH HHS/United States
- R01 AI060466/AI/NIAID NIH HHS/United States
- T32 GM007445/GM/NIGMS NIH HHS/United States
- R01 NS077869/NS/NINDS NIH HHS/United States
- R01 AI043222/AI/NIAID NIH HHS/United States
- P40 OD013117/OD/NIH HHS/United States
- R56 AI118753/AI/NIAID NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
