

Protective Effects of Euthyroidism Restoration on Mitochondria Function and Quality Control in Cardiac Pathophysiology
- PMID: 31295805
- PMCID: PMC6678270
- DOI: 10.3390/ijms20143377
Protective Effects of Euthyroidism Restoration on Mitochondria Function and Quality Control in Cardiac Pathophysiology
Abstract
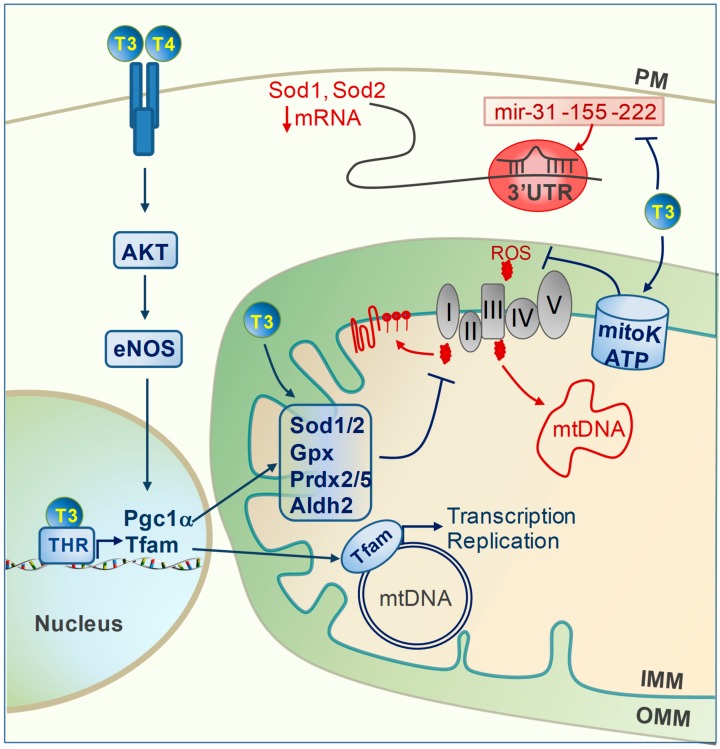
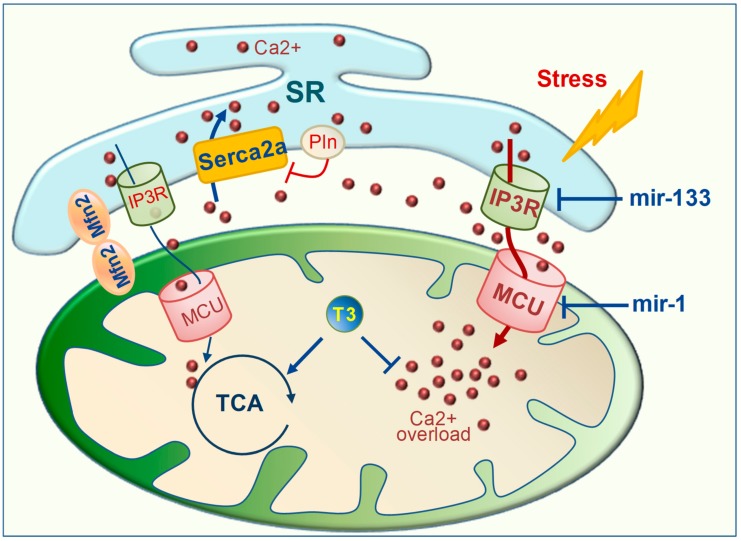
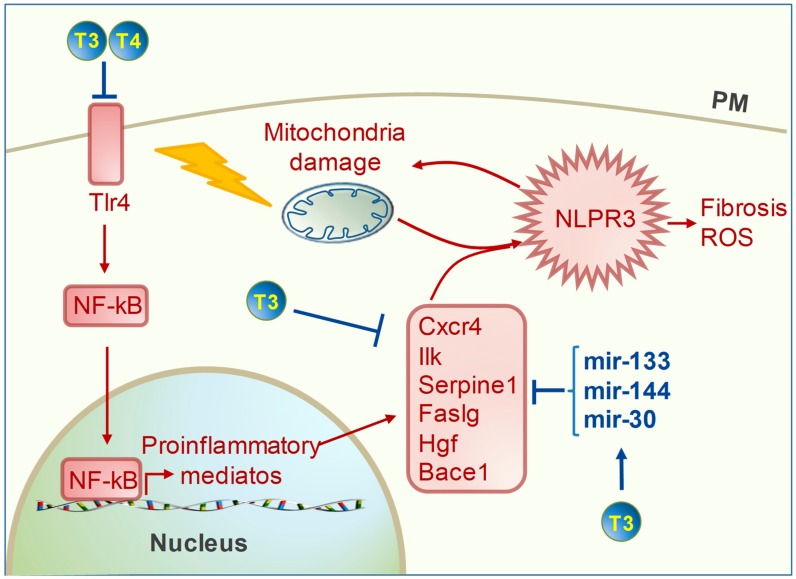
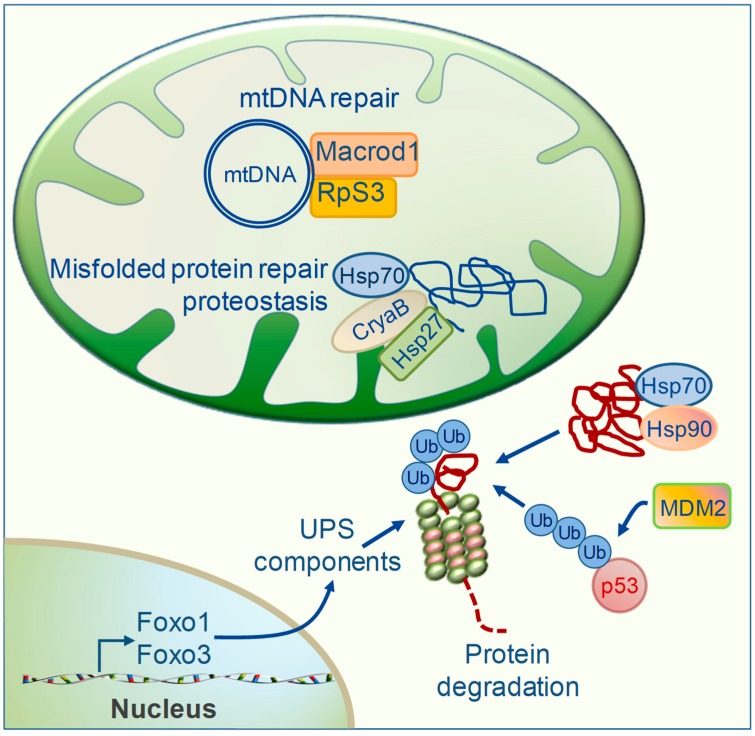
Mitochondrial dysfunctions are major contributors to heart disease onset and progression. Under ischemic injuries or cardiac overload, mitochondrial-derived oxidative stress, Ca2+ dis-homeostasis, and inflammation initiate cross-talking vicious cycles leading to defects of mitochondrial DNA, lipids, and proteins, concurrently resulting in fatal energy crisis and cell loss. Blunting such noxious stimuli and preserving mitochondrial homeostasis are essential to cell survival. In this context, mitochondrial quality control (MQC) represents an expanding research topic and therapeutic target in the field of cardiac physiology. MQC is a multi-tier surveillance system operating at the protein, organelle, and cell level to repair or eliminate damaged mitochondrial components and replace them by biogenesis. Novel evidence highlights the critical role of thyroid hormones (TH) in regulating multiple aspects of MQC, resulting in increased organelle turnover, improved mitochondrial bioenergetics, and the retention of cell function. In the present review, these emerging protective effects are discussed in the context of cardiac ischemia-reperfusion (IR) and heart failure, focusing on MQC as a strategy to blunt the propagation of connected dangerous signaling cascades and limit adverse remodeling. A better understanding of such TH-dependent signaling could provide insights into the development of mitochondria-targeted treatments in patients with cardiac disease.
Keywords: calcium handling; cardiac disease; inflammation; mitochondrial quality control; oxidative stress; thyroid hormone homeostasis.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
