

Systematic evaluation of RNA-Seq preparation protocol performance
- PMID: 31296163
- PMCID: PMC6625085
- DOI: 10.1186/s12864-019-5953-1
Systematic evaluation of RNA-Seq preparation protocol performance
Abstract
Background: RNA-Seq is currently the most widely used tool to analyze whole-transcriptome profiles. There are numerous commercial kits available to facilitate preparing RNA-Seq libraries; however, it is still not clear how some of these kits perform in terms of: 1) ribosomal RNA removal; 2) read coverage or recovery of exonic vs. intronic sequences; 3) identification of differentially expressed genes (DEGs); and 4) detection of long non-coding RNA (lncRNA). In RNA-Seq analysis, understanding the strengths and limitations of commonly used RNA-Seq library preparation protocols is important, as this technology remains costly and time-consuming.
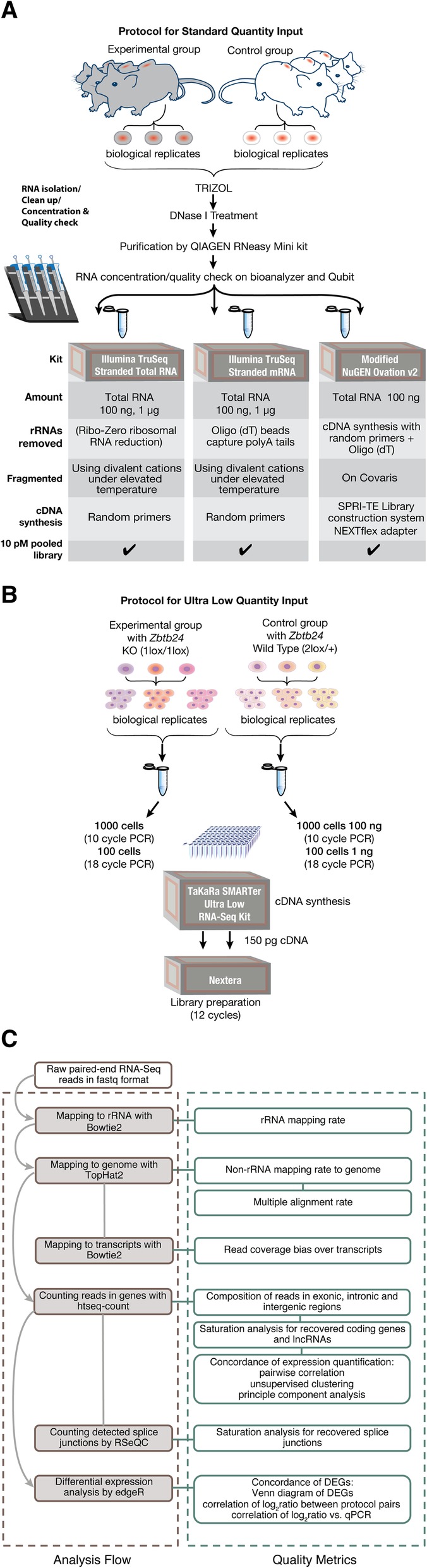
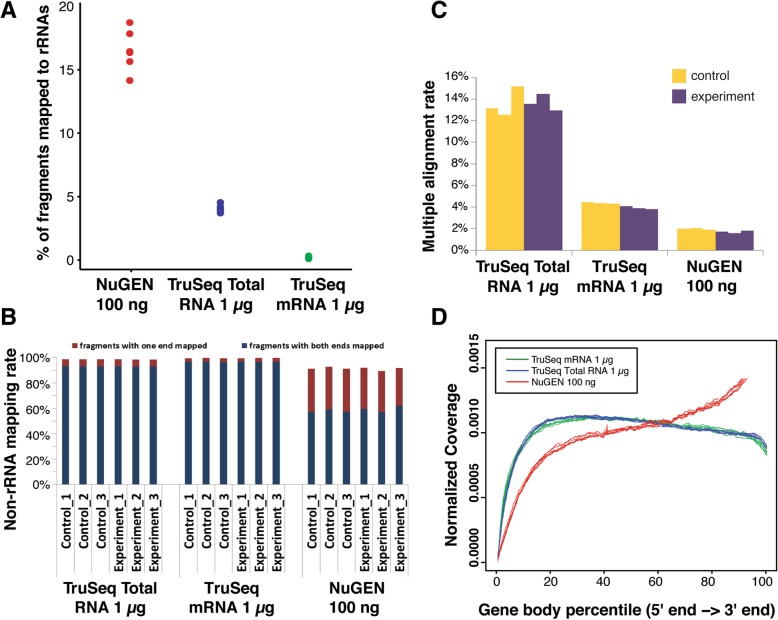
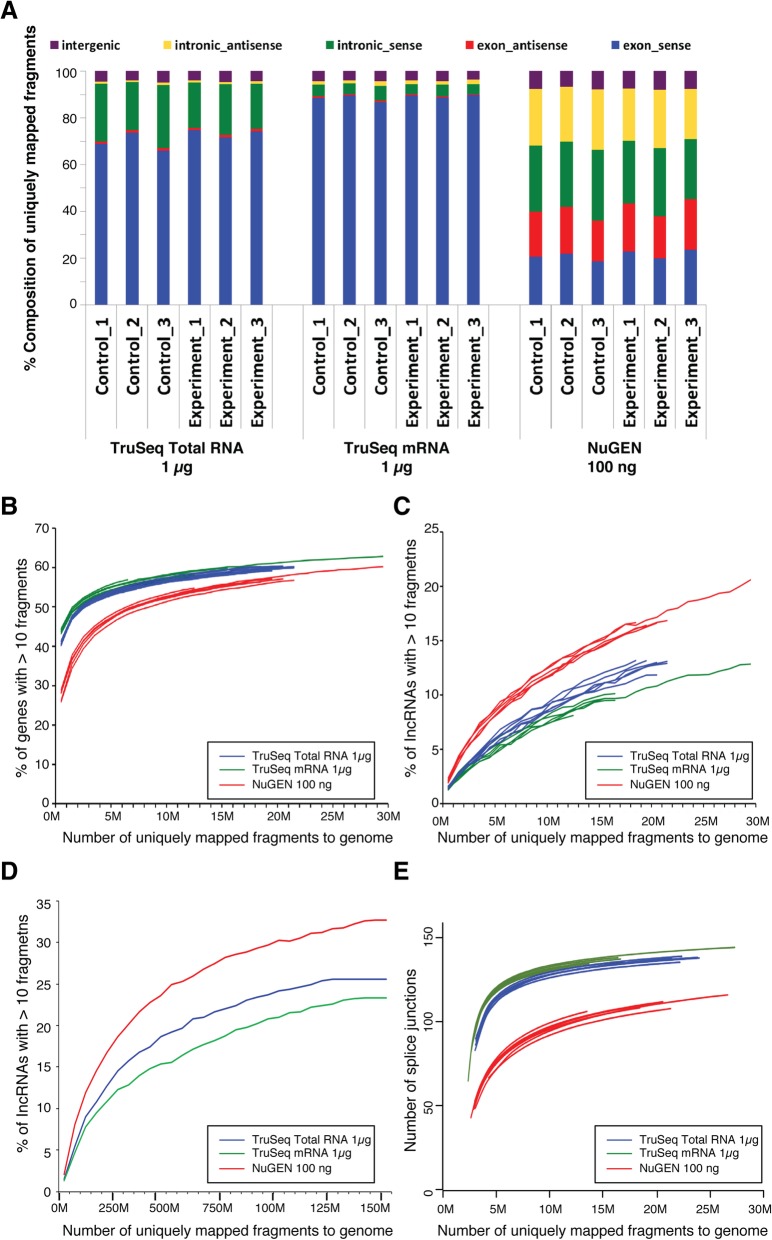
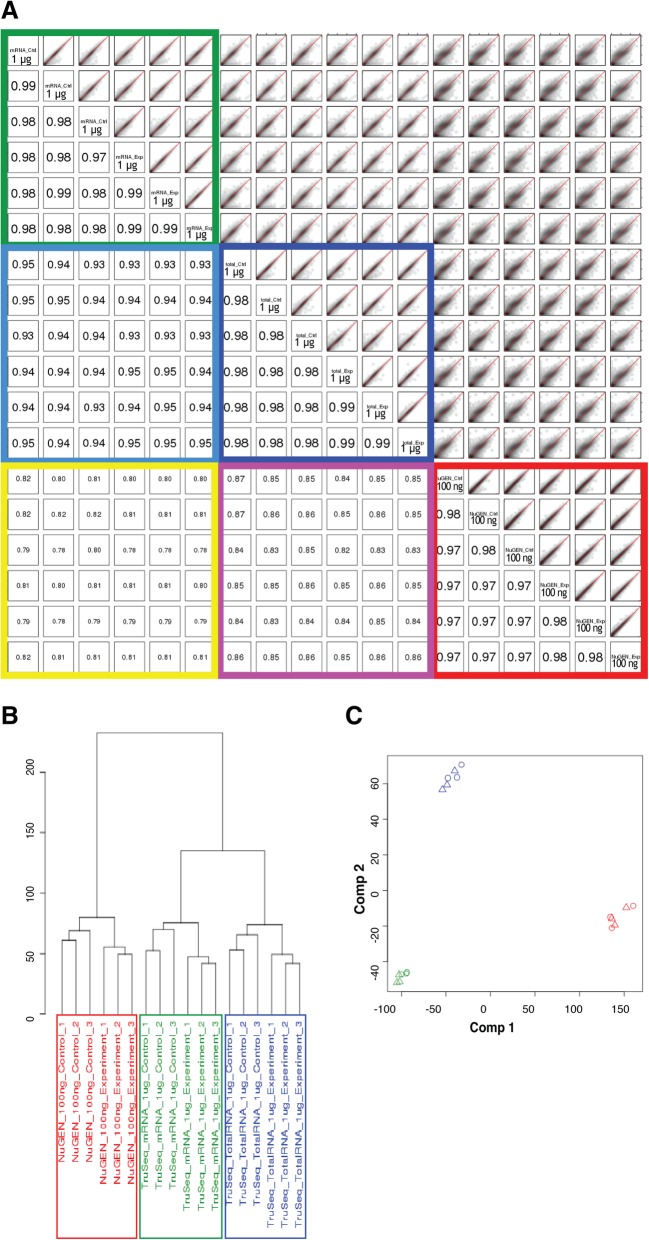
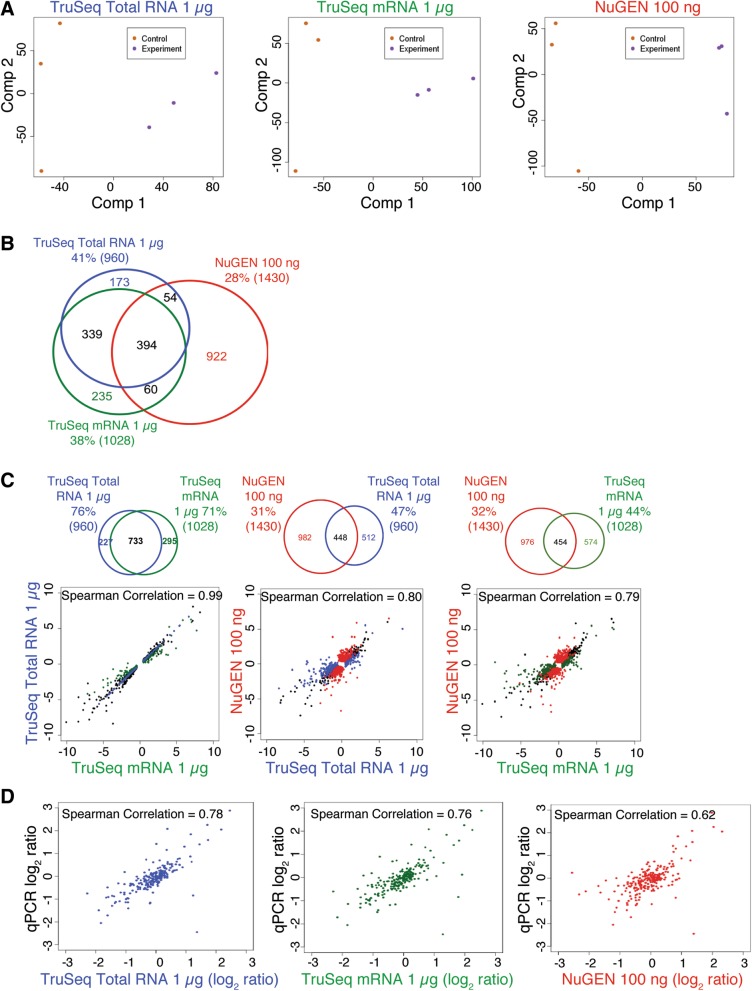
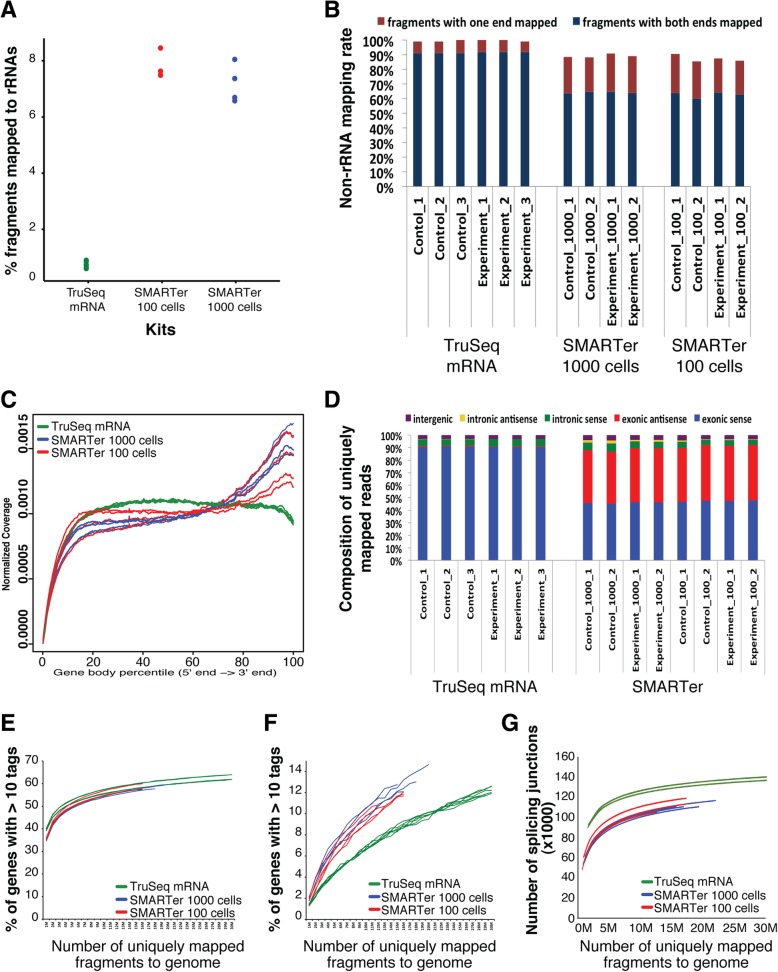
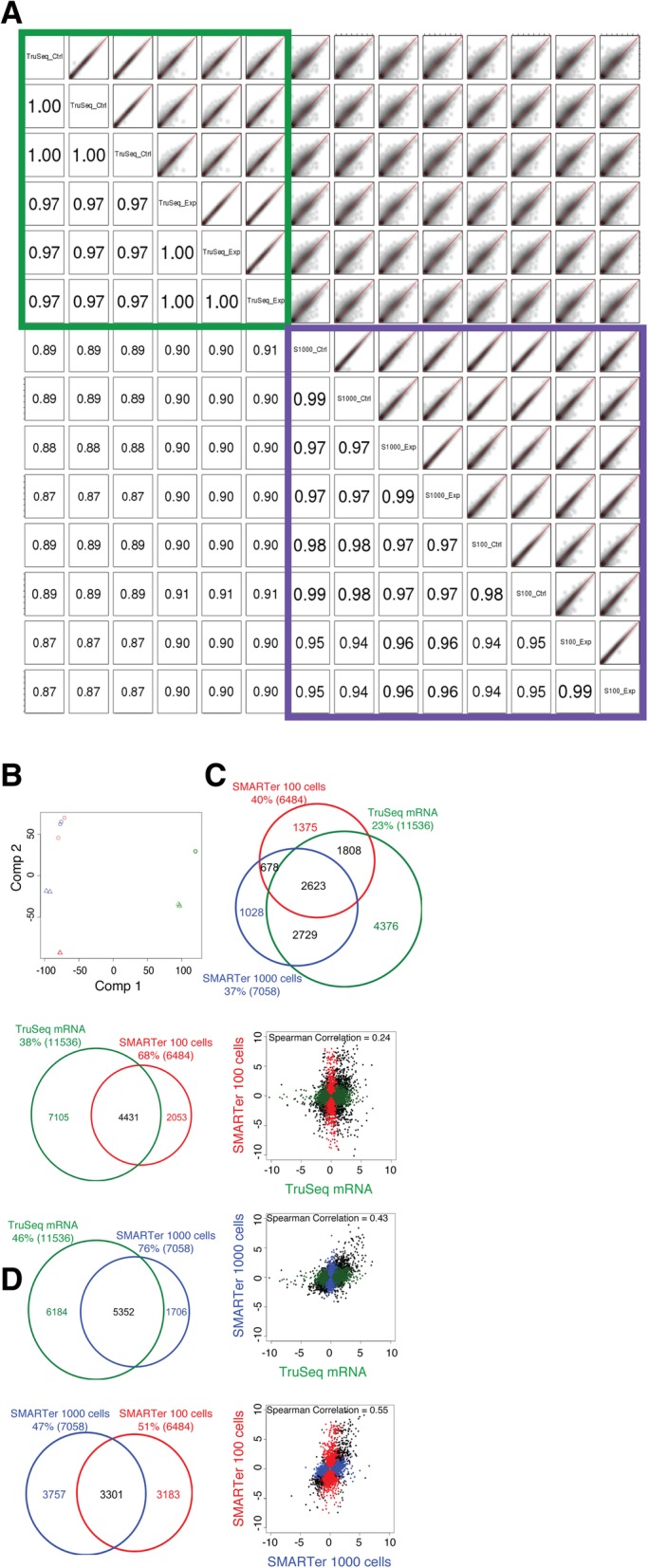
Results: In this study, we present a comprehensive evaluation of four RNA-Seq kits. We used three standard input protocols: Illumina TruSeq Stranded Total RNA and mRNA kits, a modified NuGEN Ovation v2 kit, and the TaKaRa SMARTer Ultra Low RNA Kit v3. Our evaluation of these kits included quality control measures such as overall reproducibility, 5' and 3' end-bias, and the identification of DEGs, lncRNAs, and alternatively spliced transcripts. Overall, we found that the two Illumina kits were most similar in terms of recovering DEGs, and the Illumina, modified NuGEN, and TaKaRa kits allowed identification of a similar set of DEGs. However, we also discovered that the Illumina, NuGEN and TaKaRa kits each enriched for different sets of genes.
Conclusions: At the manufacturers' recommended input RNA levels, all the RNA-Seq library preparation protocols evaluated were suitable for distinguishing between experimental groups, and the TruSeq Stranded mRNA kit was universally applicable to studies focusing on protein-coding gene profiles. The TruSeq protocols tended to capture genes with higher expression and GC content, whereas the modified NuGEN protocol tended to capture longer genes. The SMARTer Ultra Low RNA Kit may be a good choice at the low RNA input level, although it was inferior to the TruSeq mRNA kit at standard input level in terms of rRNA removal, exonic mapping rates and recovered DEGs. Therefore, the choice of RNA-Seq library preparation kit can profoundly affect data outcomes. Consequently, it is a pivotal parameter to consider when designing an RNA-Seq experiment.
Keywords: Next generation sequencing; Quality control; RNA-Seq.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
