

Topological integration of RPPA proteomic data with multi-omics data for survival prediction in breast cancer via pathway activity inference
- PMID: 31296204
- PMCID: PMC6624183
- DOI: 10.1186/s12920-019-0511-x
Topological integration of RPPA proteomic data with multi-omics data for survival prediction in breast cancer via pathway activity inference
Abstract
Background: The analysis of integrated multi-omics data enables the identification of disease-related biomarkers that cannot be identified from a single omics profile. Although protein-level data reflects the cellular status of cancer tissue more directly than gene-level data, past studies have mainly focused on multi-omics integration using gene-level data as opposed to protein-level data. However, the use of protein-level data (such as mass spectrometry) in multi-omics integration has some limitations. For example, the correlation between the characteristics of gene-level data (such as mRNA) and protein-level data is weak, and it is difficult to detect low-abundance signaling proteins that are used to target cancer. The reverse phase protein array (RPPA) is a highly sensitive antibody-based quantification method for signaling proteins. However, the number of protein features in RPPA data is extremely low compared to the number of gene features in gene-level data. In this study, we present a new method for integrating RPPA profiles with RNA-Seq and DNA methylation profiles for survival prediction based on the integrative directed random walk (iDRW) framework proposed in our previous study. In the iDRW framework, each omics profile is merged into a single pathway profile that reflects the topological information of the pathway. In order to address the sparsity of RPPA profiles, we employ the random walk with restart (RWR) approach on the pathway network.
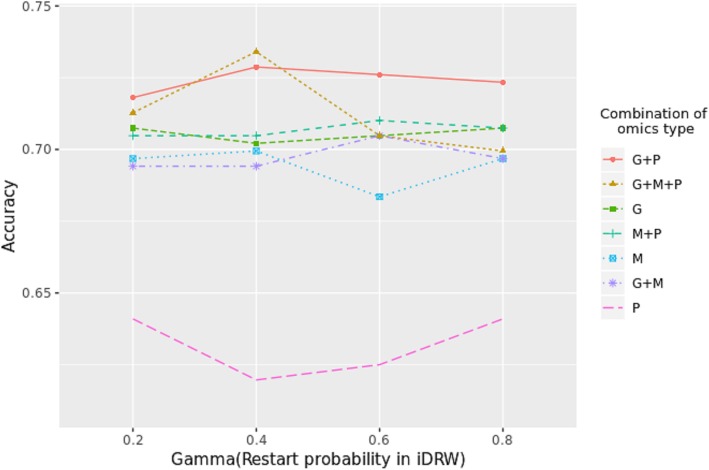
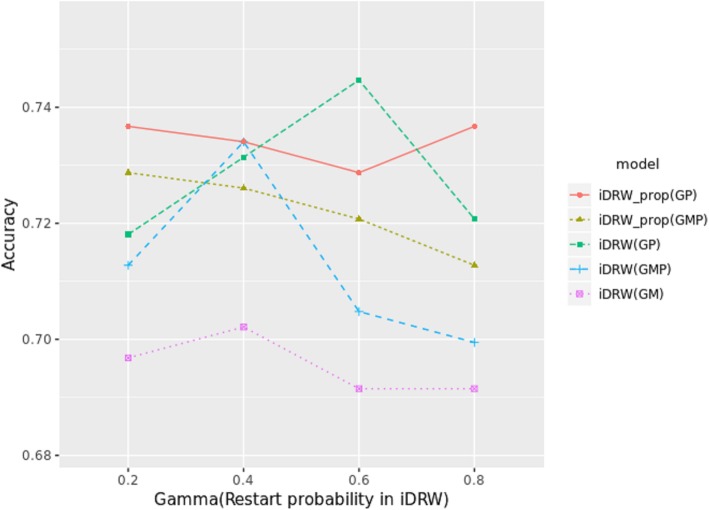
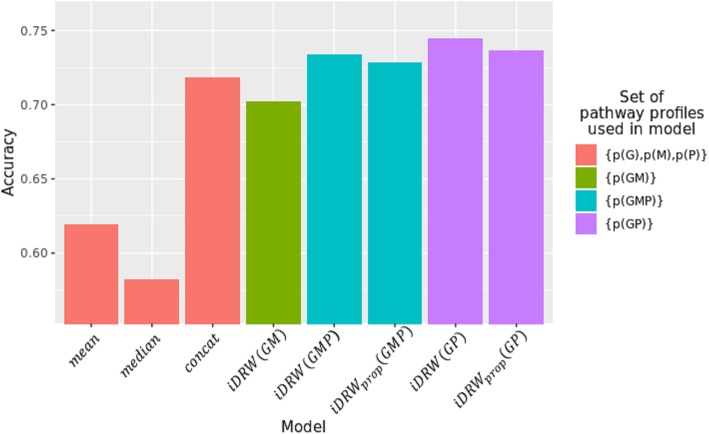
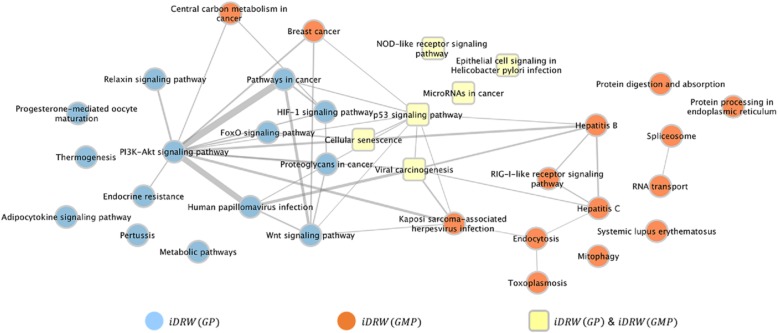
Results: Our model was validated using survival prediction analysis for a breast cancer dataset from The Cancer Genome Atlas. Our proposed model exhibited improved performance compared with other methods that utilize pathway information and also out-performed models that did not include the RPPA data utilized in our study. The risk pathways identified for breast cancer in this study were closely related to well-known breast cancer risk pathways.
Conclusions: Our results indicated that RPPA data is useful for survival prediction for breast cancer patients under our framework. We also observed that iDRW effectively integrates RNA-Seq, DNA methylation, and RPPA profiles, while variation in the composition of the omics data can affect both prediction performance and risk pathway identification. These results suggest that omics data composition is a critical parameter for iDRW.
Keywords: Breast cancer; Integrative analysis; Multi-omics data; Network propagation; Pathway-based analysis; Random walk; Reverse phase protein Array; Survival prediction.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
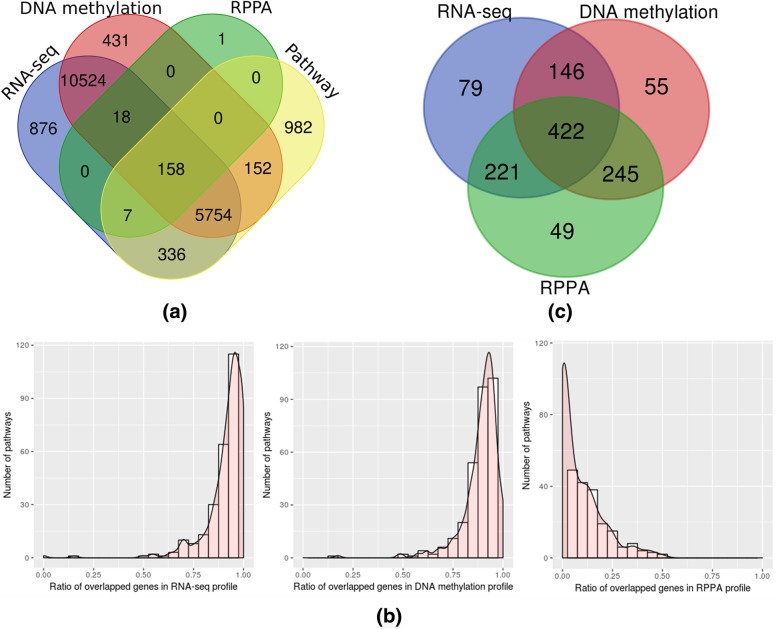
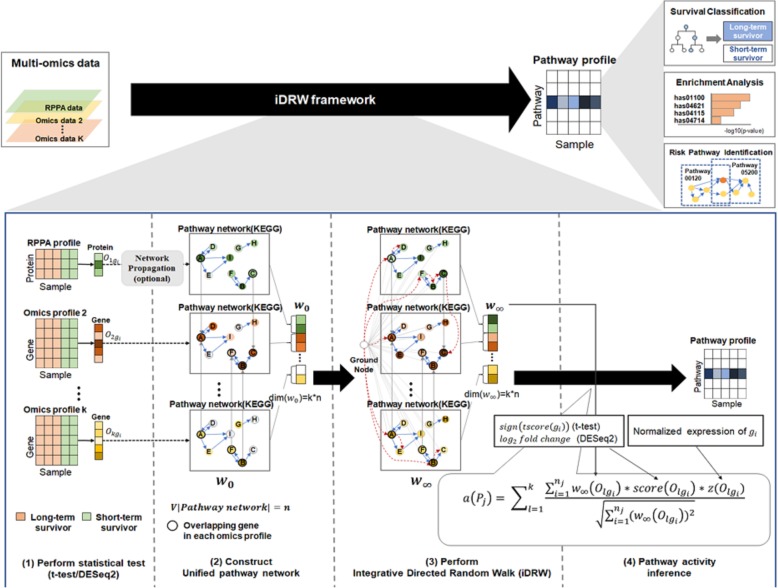
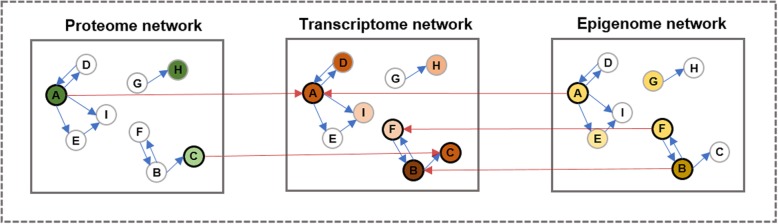
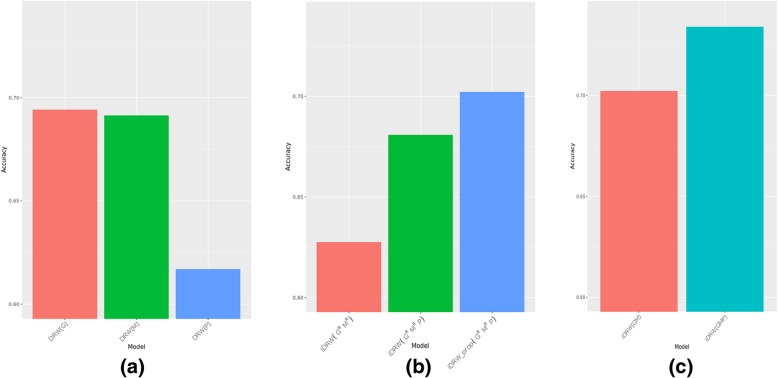
Similar articles
-
Robust pathway-based multi-omics data integration using directed random walks for survival prediction in multiple cancer studies.Biol Direct. 2019 Apr 29;14(1):8. doi: 10.1186/s13062-019-0239-8. Biol Direct. 2019. PMID: 31036036 Free PMC article.
-
Integrative pathway-based survival prediction utilizing the interaction between gene expression and DNA methylation in breast cancer.BMC Med Genomics. 2018 Sep 14;11(Suppl 3):68. doi: 10.1186/s12920-018-0389-z. BMC Med Genomics. 2018. PMID: 30255812 Free PMC article.
-
Predicting censored survival data based on the interactions between meta-dimensional omics data in breast cancer.J Biomed Inform. 2015 Aug;56:220-8. doi: 10.1016/j.jbi.2015.05.019. Epub 2015 Jun 3. J Biomed Inform. 2015. PMID: 26048077 Free PMC article.
-
Reverse phase protein arrays in signaling pathways: a data integration perspective.Drug Des Devel Ther. 2015 Jul 7;9:3519-27. doi: 10.2147/DDDT.S38375. eCollection 2015. Drug Des Devel Ther. 2015. PMID: 26185419 Free PMC article. Review.
-
Subtyping of breast cancer using reverse phase protein arrays.Expert Rev Proteomics. 2014 Dec;11(6):757-70. doi: 10.1586/14789450.2014.971113. Expert Rev Proteomics. 2014. PMID: 25400094 Review.
Cited by
-
Deepening into Intracellular Signaling Landscape through Integrative Spatial Proteomics and Transcriptomics in a Lymphoma Model.Biomolecules. 2021 Nov 26;11(12):1776. doi: 10.3390/biom11121776. Biomolecules. 2021. PMID: 34944421 Free PMC article.
-
Overview of methods for characterization and visualization of a protein-protein interaction network in a multi-omics integration context.Front Mol Biosci. 2022 Sep 8;9:962799. doi: 10.3389/fmolb.2022.962799. eCollection 2022. Front Mol Biosci. 2022. PMID: 36158572 Free PMC article. Review.
-
Visible neural networks for multi-omics integration: a critical review.Front Artif Intell. 2025 Jul 17;8:1595291. doi: 10.3389/frai.2025.1595291. eCollection 2025. Front Artif Intell. 2025. PMID: 40746431 Free PMC article.
References
-
- Joyce AR, Palsson BØ. The model organism as a system: integrating'omics' data sets. Nat Rev Mol Cell Biol. 2006;7(3):198. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
