

Regulation of tumor angiogenesis and mesenchymal-endothelial transition by p38α through TGF-β and JNK signaling
- PMID: 31296856
- PMCID: PMC6624205
- DOI: 10.1038/s41467-019-10946-y
Regulation of tumor angiogenesis and mesenchymal-endothelial transition by p38α through TGF-β and JNK signaling
Abstract
The formation of new blood vessels is essential for normal development, tissue repair and tumor growth. Here we show that inhibition of the kinase p38α enhances angiogenesis in human and mouse colon tumors. Mesenchymal cells can contribute to tumor angiogenesis by regulating proliferation and migration of endothelial cells. We show that p38α negatively regulates an angiogenic program in mesenchymal stem/stromal cells (MSCs), multipotent progenitors found in perivascular locations. This program includes the acquisition of an endothelial phenotype by MSCs mediated by both TGF-β and JNK, and negatively regulated by p38α. Abrogation of p38α in mesenchymal cells increases tumorigenesis, which correlates with enhanced angiogenesis. Using genetic models, we show that p38α regulates the acquisition of an endothelial-like phenotype by mesenchymal cells in colon tumors and damage tissue. Taken together, our results indicate that p38α in mesenchymal cells restrains a TGF-β-induced angiogenesis program including their ability to transdifferentiate into endothelial cells.
Conflict of interest statement
The authors declare no competing interests.
Figures

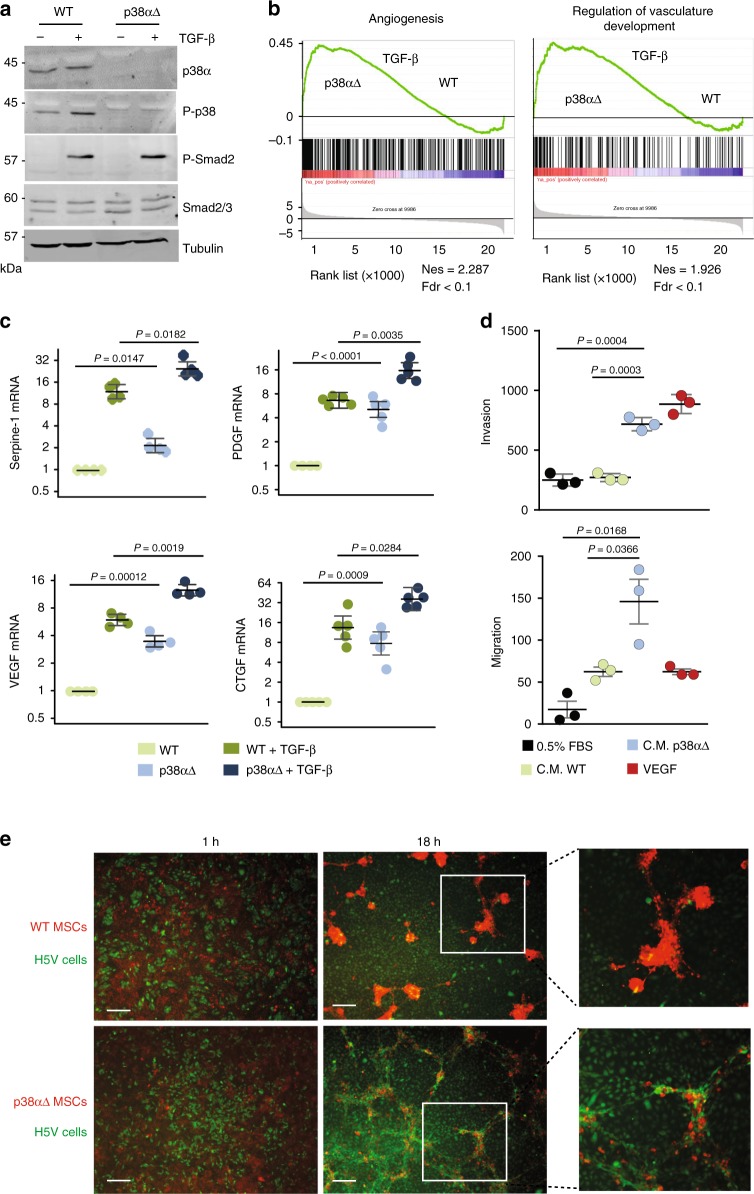




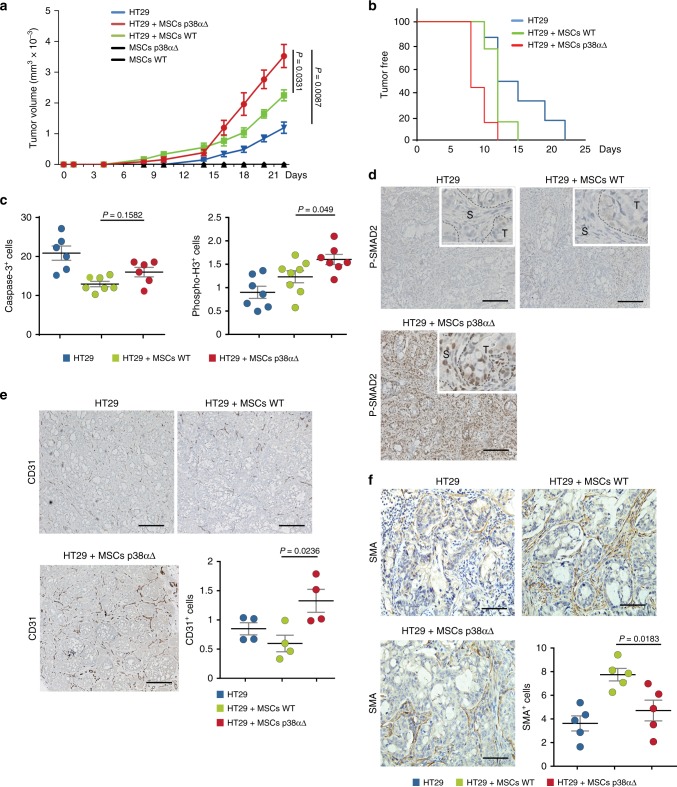


References
Publication types
MeSH terms
Substances
Grants and funding
- ERC 294665/EC | EU Framework Programme for Research and Innovation H2020 | H2020 Priority Excellent Science | H2020 European Research Council (H2020 Excellent Science - European Research Council)/International
- SAF2016-81043-R/Ministerio de Economía y Competitividad (Ministry of Economy and Competitiveness)/International
- 2017 SGR-557/Departament d'Innovació, Universitats i Empresa, Generalitat de Catalunya (Department of Innovation, Education and Enterprise, Government of Catalonia)/International
- 20133430/Fundació la Marató de TV3 (TV3 Marathon Foundation)/International
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
