

Apabetalone (RVX-208) reduces vascular inflammation in vitro and in CVD patients by a BET-dependent epigenetic mechanism
- PMID: 31300040
- PMCID: PMC6626370
- DOI: 10.1186/s13148-019-0696-z
Apabetalone (RVX-208) reduces vascular inflammation in vitro and in CVD patients by a BET-dependent epigenetic mechanism
Abstract
Background: Apabetalone (RVX-208) is a bromodomain and extraterminal protein inhibitor (BETi) that in phase II trials reduced the relative risk (RR) of major adverse cardiac events (MACE) in patients with cardiovascular disease (CVD) by 44% and in diabetic CVD patients by 57% on top of statins. A phase III trial, BETonMACE, is currently assessing apabetalone's ability to reduce MACE in statin-treated post-acute coronary syndrome type 2 diabetic CVD patients with low high-density lipoprotein C. The leading cause of MACE is atherosclerosis, driven by dysfunctional lipid metabolism and chronic vascular inflammation (VI). In vitro studies have implicated the BET protein BRD4 as an epigenetic driver of inflammation and atherogenesis, suggesting that BETi may be clinically effective in combating VI. Here, we assessed apabetalone's ability to regulate inflammation-driven gene expression and cell adhesion in vitro and investigated the mechanism by which apabetalone suppresses expression. The clinical impact of apabetalone on mediators of VI was assessed with proteomic analysis of phase II CVD patient plasma.
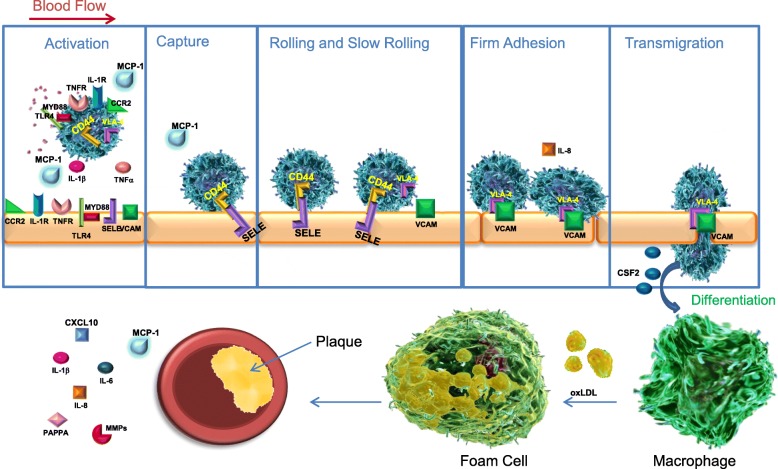
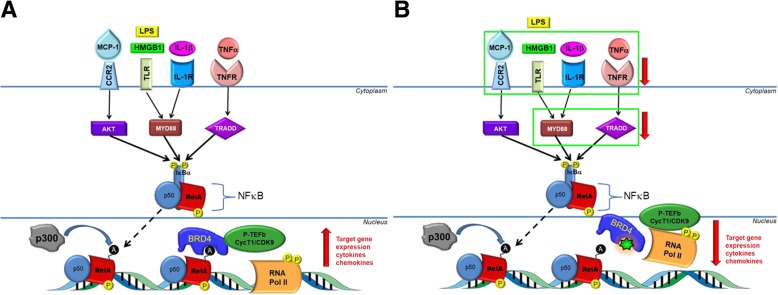
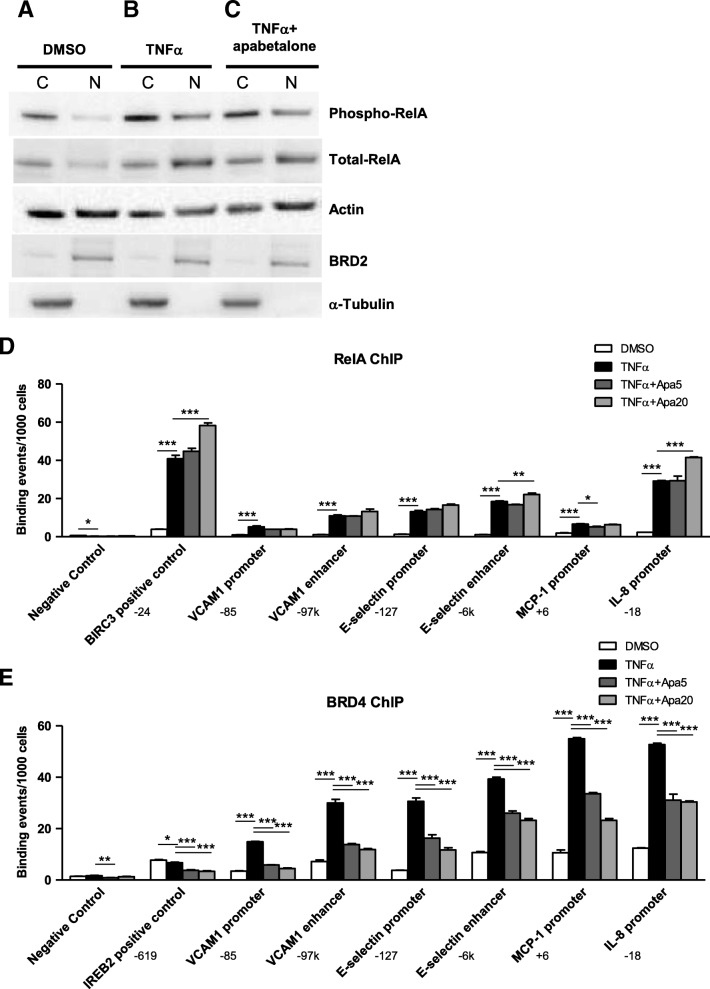
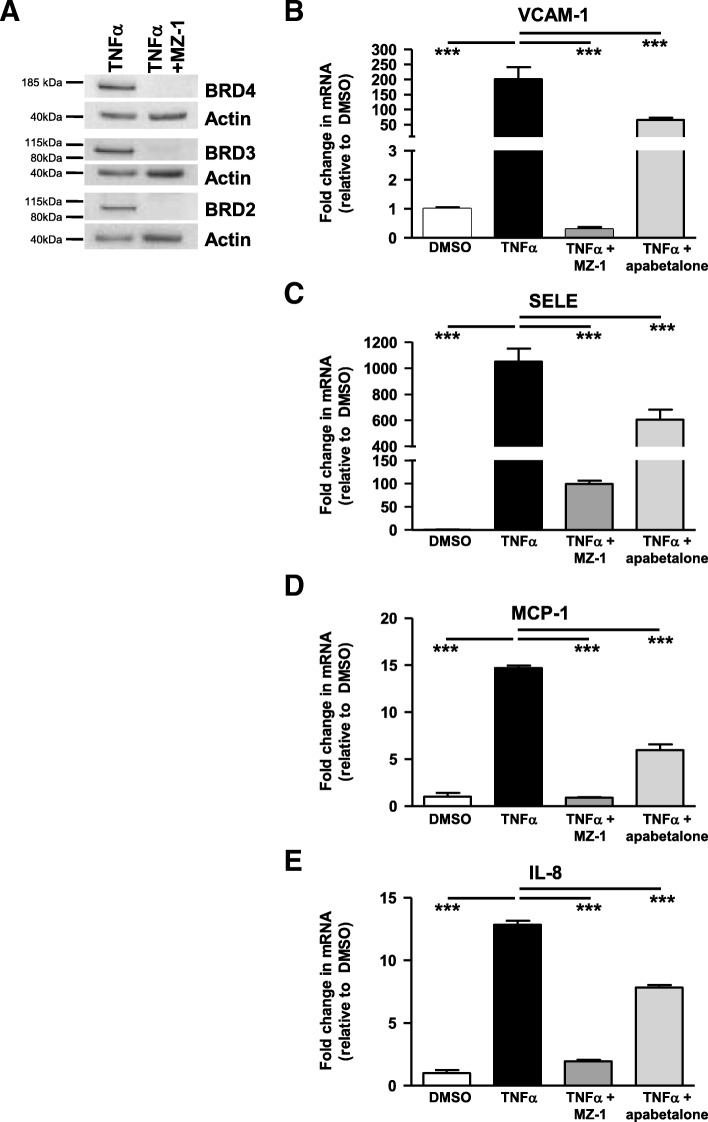
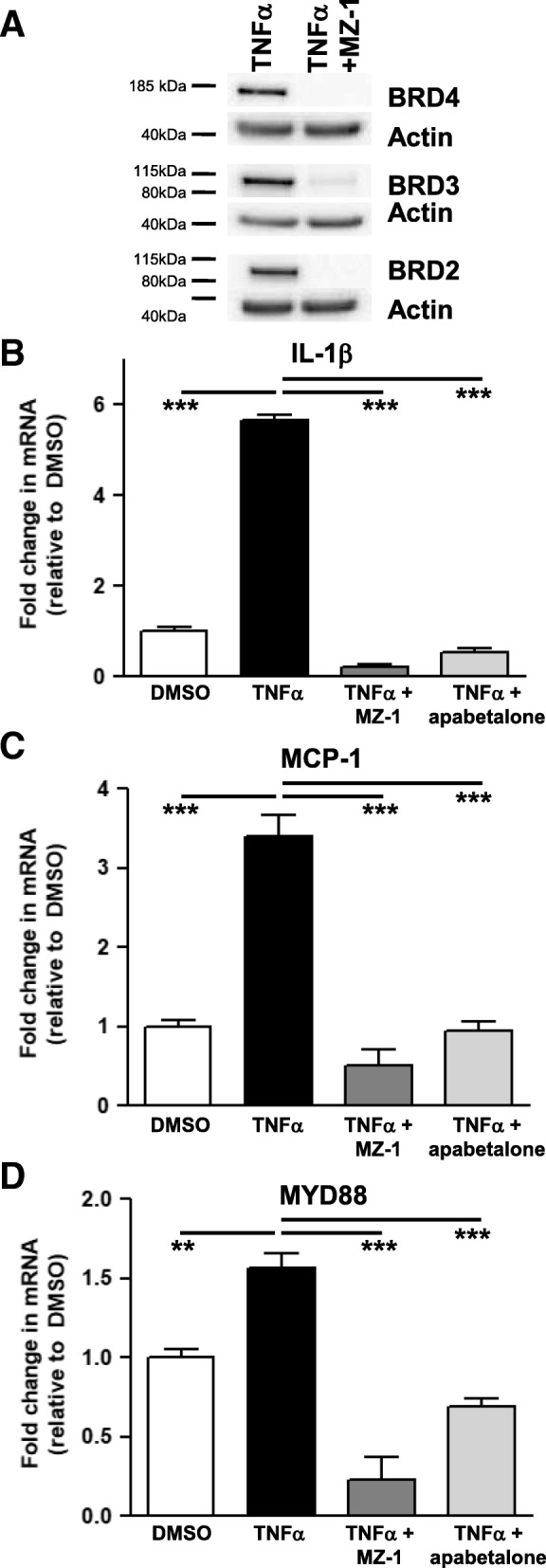
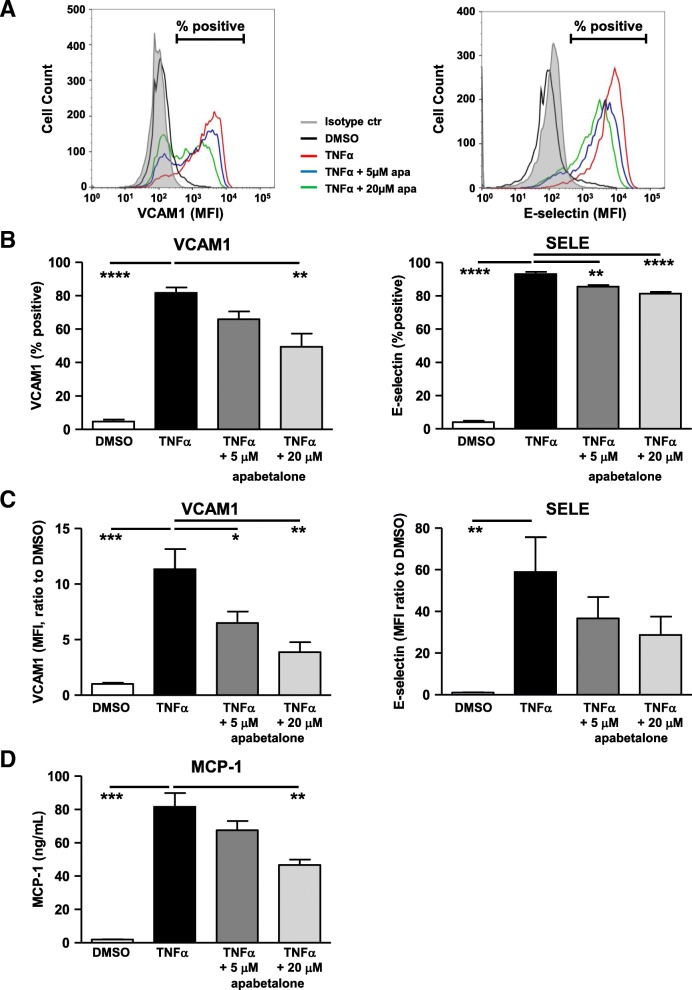
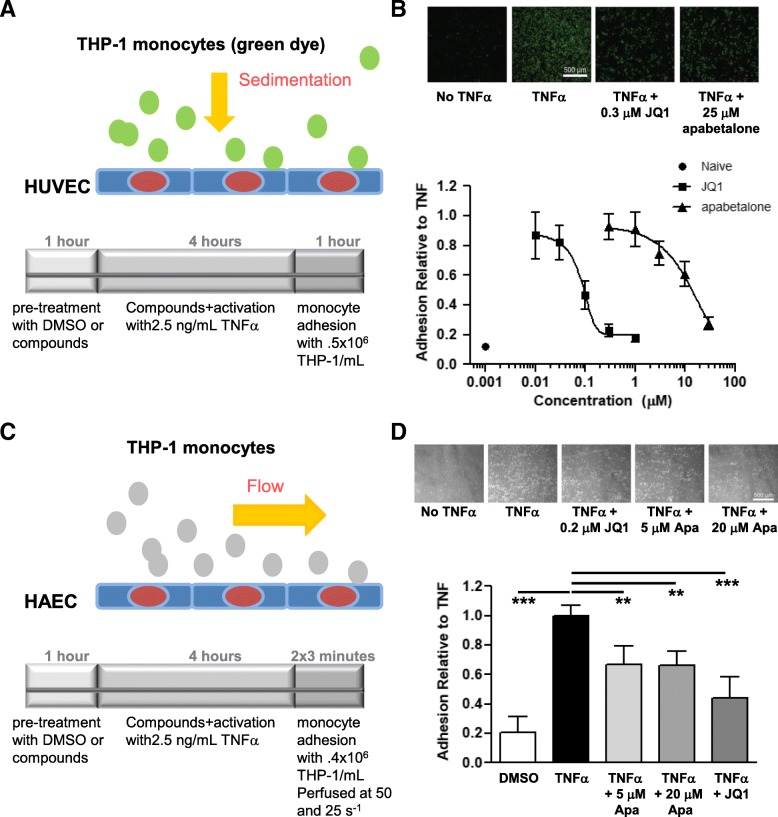
Results: In vitro, apabetalone prevented inflammatory (TNFα, LPS, or IL-1β) induction of key factors that drive endothelial activation, monocyte recruitment, adhesion, and plaque destabilization. BRD4 abundance on inflammatory and adhesion gene promoters and enhancers was reduced by apabetalone. BRD2-4 degradation by MZ-1 also prevented TNFα-induced transcription of monocyte and endothelial cell adhesion molecules and inflammatory mediators, confirming BET-dependent regulation. Transcriptional regulation by apabetalone translated into a reduction in monocyte adhesion to an endothelial monolayer. In a phase II trial, apabetalone treatment reduced the abundance of multiple VI mediators in the plasma of CVD patients (SOMAscan® 1.3 k). These proteins correlate with CVD risk and include adhesion molecules, cytokines, and metalloproteinases. Ingenuity® Pathway Analysis (IPA®) predicted that apabetalone inhibits pro-atherogenic regulators and pathways and prevents disease states arising from leukocyte recruitment.
Conclusions: Apabetalone suppressed gene expression of VI mediators in monocytes and endothelial cells by inhibiting BET-dependent transcription induced by multiple inflammatory stimuli. In CVD patients, apabetalone treatment reduced circulating levels of VI mediators, an outcome conducive with atherosclerotic plaque stabilization and MACE reduction. Inhibition of inflammatory and adhesion molecule gene expression by apabetalone is predicted to contribute to MACE reduction in the phase III BETonMACE trial.
Keywords: Adhesion; Apabetalone; Atherosclerosis; BRD4; Bromodomain; CVD; Diabetes; Epigenetics; HUVEC; THP-1 monocytes; Vascular inflammation; endothelium.
Conflict of interest statement
All authors that are Resverlogix employees receive salaries and shares from the company.
Figures
References
-
- World Heath Organization. Cardiovascular diseases (CVDs) [Webpage]. 2017. https://www.who.int/en/news-room/fact-sheets/detail/cardiovascular-disea.... [updated May 17, 2017; cited 2019 January 10, 2019].
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
