

AMPA receptor GluA2 subunit defects are a cause of neurodevelopmental disorders
- PMID: 31300657
- PMCID: PMC6626132
- DOI: 10.1038/s41467-019-10910-w
AMPA receptor GluA2 subunit defects are a cause of neurodevelopmental disorders
Abstract
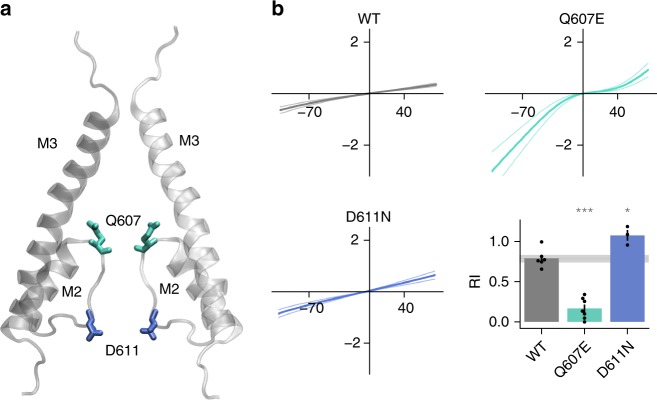
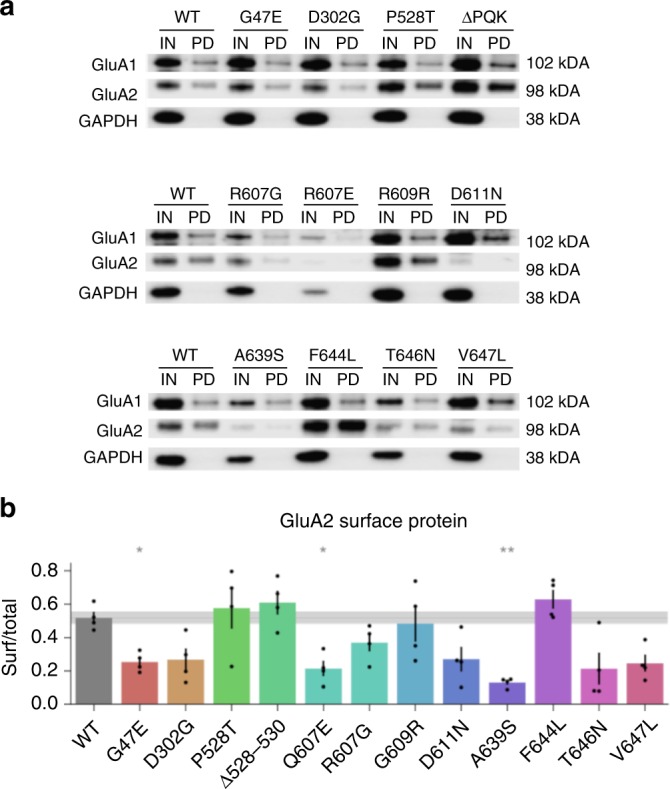
AMPA receptors (AMPARs) are tetrameric ligand-gated channels made up of combinations of GluA1-4 subunits encoded by GRIA1-4 genes. GluA2 has an especially important role because, following post-transcriptional editing at the Q607 site, it renders heteromultimeric AMPARs Ca2+-impermeable, with a linear relationship between current and trans-membrane voltage. Here, we report heterozygous de novo GRIA2 mutations in 28 unrelated patients with intellectual disability (ID) and neurodevelopmental abnormalities including autism spectrum disorder (ASD), Rett syndrome-like features, and seizures or developmental epileptic encephalopathy (DEE). In functional expression studies, mutations lead to a decrease in agonist-evoked current mediated by mutant subunits compared to wild-type channels. When GluA2 subunits are co-expressed with GluA1, most GRIA2 mutations cause a decreased current amplitude and some also affect voltage rectification. Our results show that de-novo variants in GRIA2 can cause neurodevelopmental disorders, complementing evidence that other genetic causes of ID, ASD and DEE also disrupt glutamatergic synaptic transmission.
Conflict of interest statement
E.T., K.G.M., T.S.-S., R.E.P., R.W., and M.T.C. are employees of GeneDx. E.E.E. is on the scientific advisory board (SAB) of DNAnexus, Inc. The remaining authors declare no competing interests.
Figures
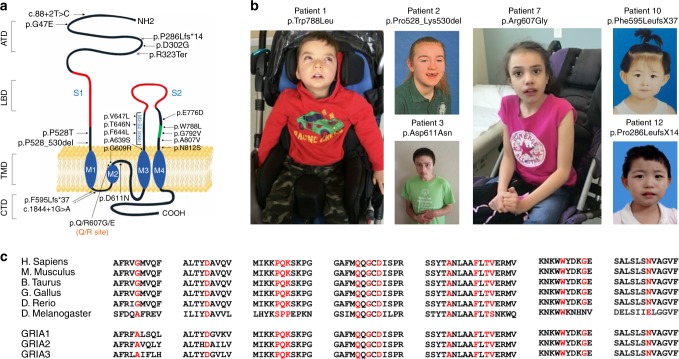
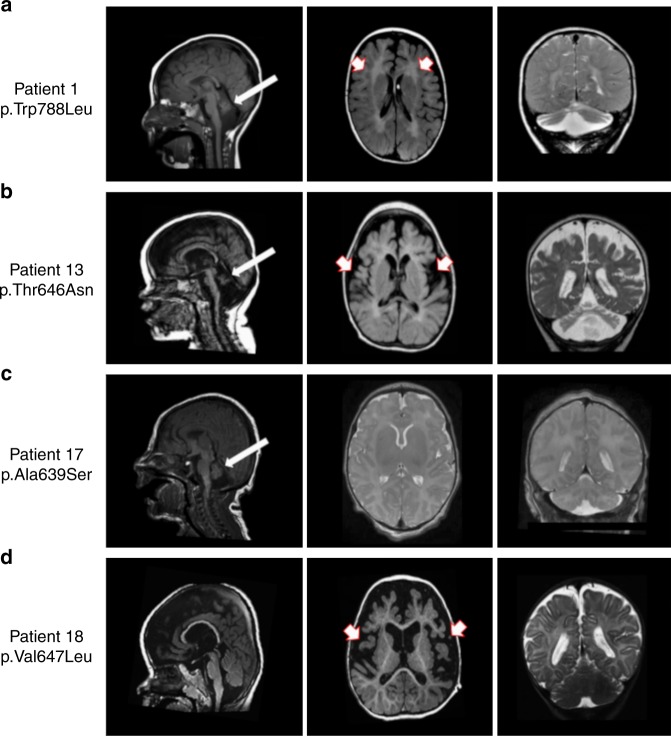
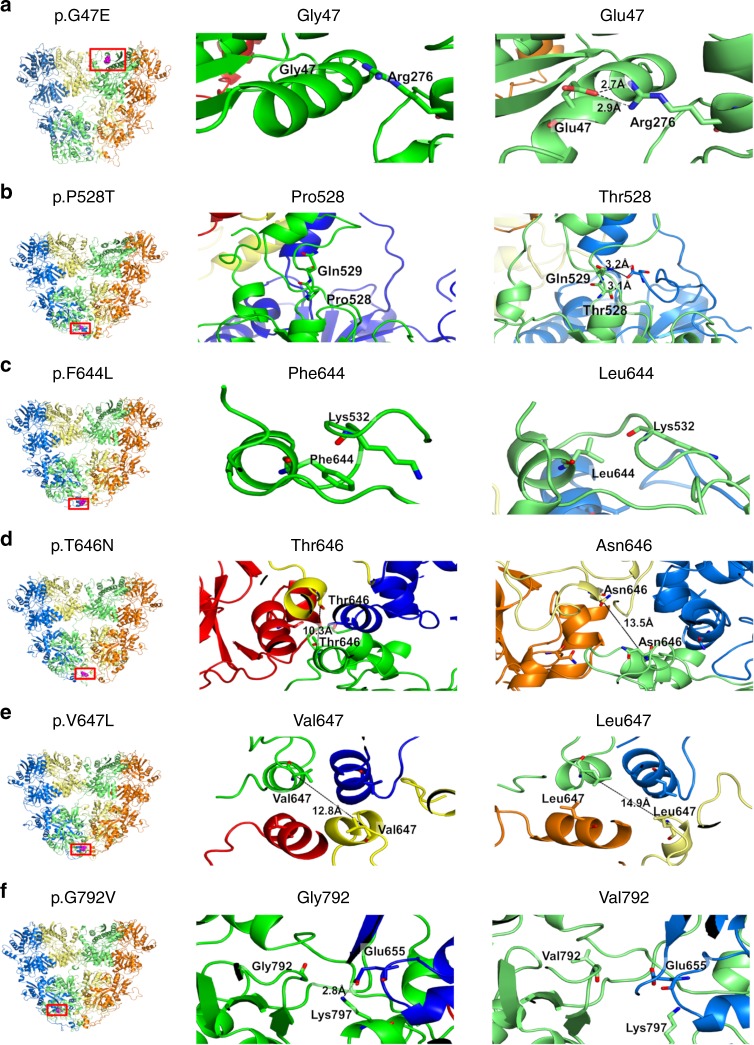
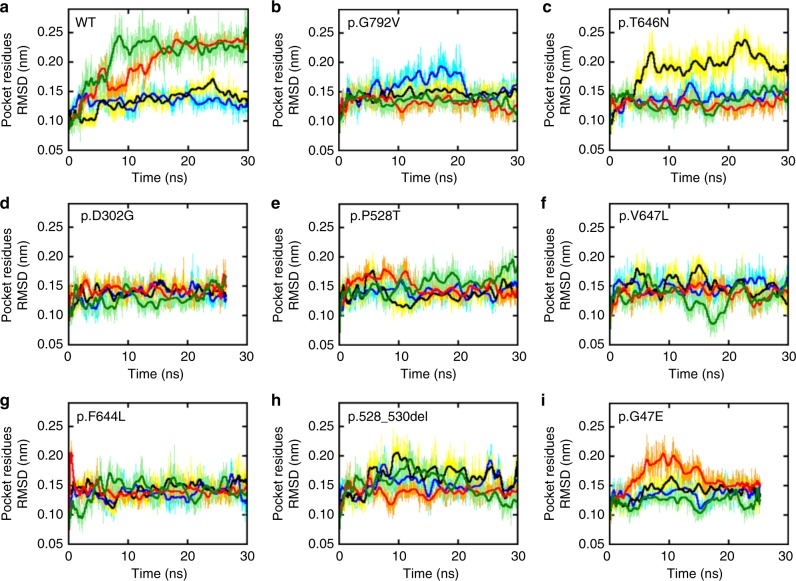



Comment in
-
Vulnerabilities in a Dominant Receptor Subunit.Epilepsy Curr. 2020 Mar;20(2):97-98. doi: 10.1177/1535759720904359. Epub 2020 Feb 17. Epilepsy Curr. 2020. PMID: 32064923 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
- G0900613/MRC_/Medical Research Council/United Kingdom
- G0400136/MRC_/Medical Research Council/United Kingdom
- G1001253/MRC_/Medical Research Council/United Kingdom
- R605/0717/DMT_/The Dunhill Medical Trust/United Kingdom
- G108/638/MRC_/Medical Research Council/United Kingdom
- G0200373/MRC_/Medical Research Council/United Kingdom
- G0600368/MRC_/Medical Research Council/United Kingdom
- G0802760/MRC_/Medical Research Council/United Kingdom
- G0601440/MRC_/Medical Research Council/United Kingdom
- MR/S006753/1/MRC_/Medical Research Council/United Kingdom
- R01 MH101221/MH/NIMH NIH HHS/United States
- G116/147/MRC_/Medical Research Council/United Kingdom
- G9805989/MRC_/Medical Research Council/United Kingdom
- U54 HD083091/HD/NICHD NIH HHS/United States
- MR/L01095X/1/MRC_/Medical Research Council/United Kingdom
- G0801316/MRC_/Medical Research Council/United Kingdom
- R01 NS069605/NS/NINDS NIH HHS/United States
- 209807/Z/17/Z/WT_/Wellcome Trust/United Kingdom
- 212285/Z/18/Z/WT_/Wellcome Trust/United Kingdom
- MR/S01165X/1/MRC_/Medical Research Council/United Kingdom
- G0400627/MRC_/Medical Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
