

Axon injury signaling and compartmentalized injury response in glaucoma
- PMID: 31301400
- PMCID: PMC6898776
- DOI: 10.1016/j.preteyeres.2019.07.002
Axon injury signaling and compartmentalized injury response in glaucoma
Abstract
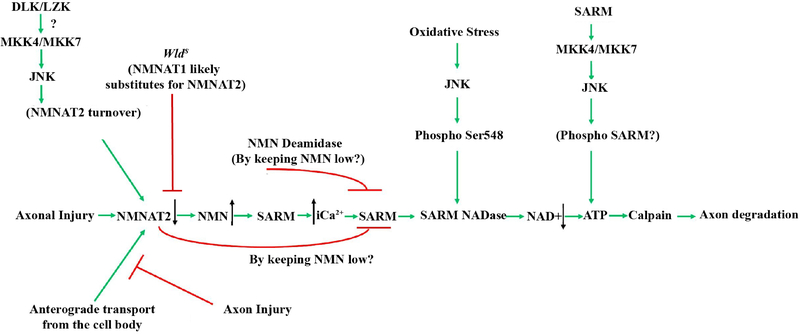
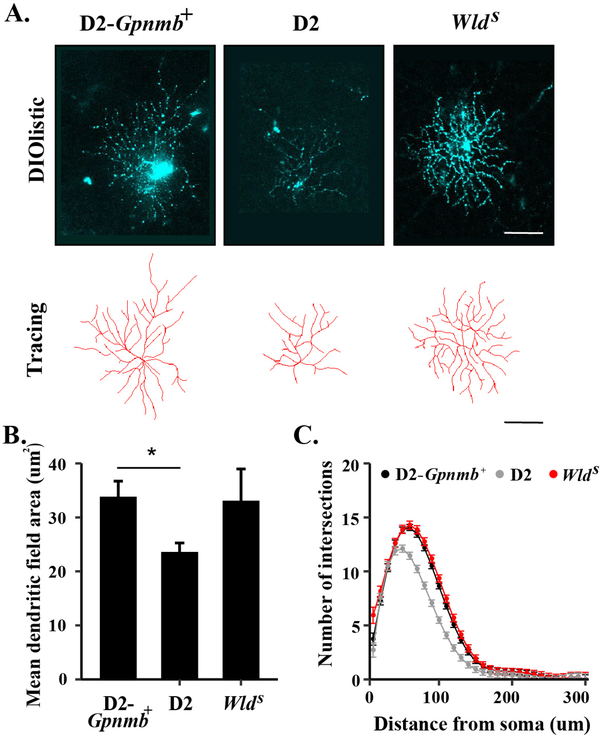
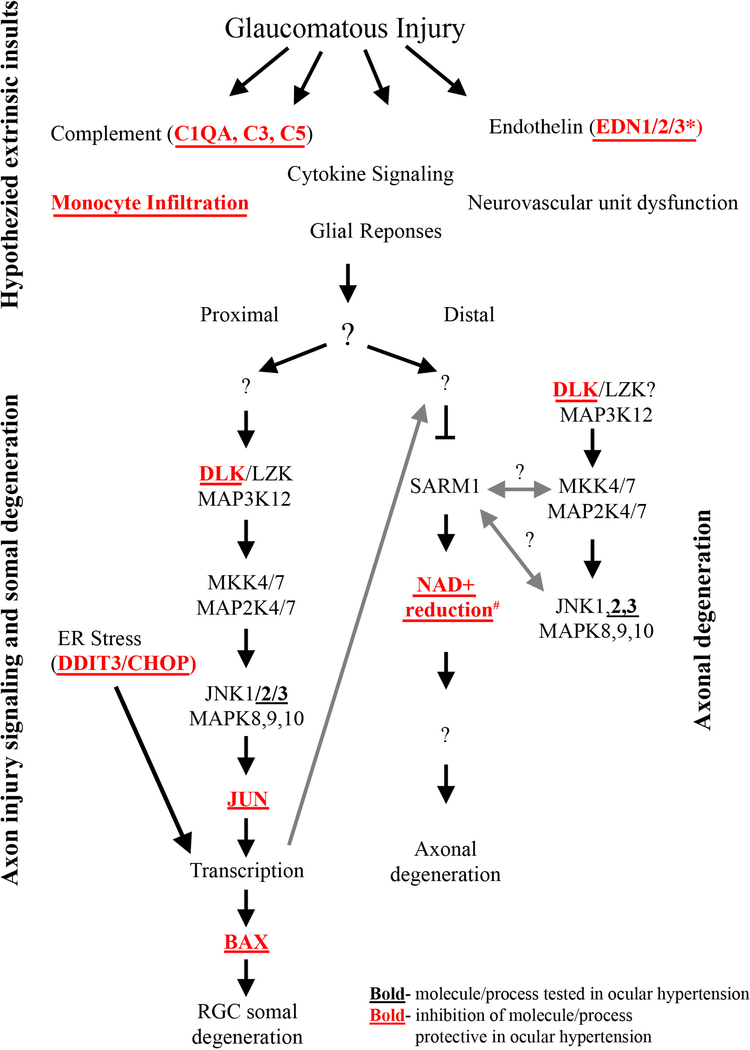
Axonal degeneration is an active, highly controlled process that contributes to beneficial processes, such as developmental pruning, but also to neurodegeneration. In glaucoma, ocular hypertension leads to vision loss by killing the output neurons of the retina, the retinal ganglion cells (RGCs). Multiple processes have been proposed to contribute to and/or mediate axonal injury in glaucoma, including: neuroinflammation, loss of neurotrophic factors, dysregulation of the neurovascular unit, and disruption of the axonal cytoskeleton. While the inciting injury to RGCs in glaucoma is complex and potentially heterogeneous, axonal injury is ultimately thought to be the key insult that drives glaucomatous neurodegeneration. Glaucomatous neurodegeneration is a complex process, with multiple molecular signals contributing to RGC somal loss and axonal degeneration. Furthermore, the propagation of the axonal injury signal is complex, with injury triggering programs of degeneration in both the somal and axonal compartment. Further complicating this process is the involvement of multiple cell types that are known to participate in the process of axonal and neuronal degeneration after glaucomatous injury. Here, we review the axonal signaling that occurs after injury and the molecular signaling programs currently known to be important for somal and axonal degeneration after glaucoma-relevant axonal injuries.
Keywords: Apoptosis; Axon; Axonopathy; Dendritic remodeling; Intraocular pressure; Neuroprotection; Optic neuropathy; Synaptic loss.
Copyright © 2019 Elsevier Ltd. All rights reserved.
Figures
References
-
- Agostinone J, Di Polo A, 2015. Retinal ganglion cell dendrite pathology and synapse loss: Implications for glaucoma. Prog Brain Res 220, 199–216. - PubMed
-
- Akhter R, Sanphui P, Das H, Saha P, Biswas SC, 2015. The regulation of p53 up-regulated modulator of apoptosis by JNK/c-Jun pathway in beta-amyloid-induced neuron death. J Neurochem 134, 1091–1103. - PubMed
-
- Almasieh M, Wilson AM, Morquette B, Cueva Vargas JL, Di Polo A, 2012. The molecular basis of retinal ganglion cell death in glaucoma. Prog Retin Eye Res 31, 152–181. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
