

Mutations in ANAPC1, Encoding a Scaffold Subunit of the Anaphase-Promoting Complex, Cause Rothmund-Thomson Syndrome Type 1
- PMID: 31303264
- PMCID: PMC6731352
- DOI: 10.1016/j.ajhg.2019.06.011
Mutations in ANAPC1, Encoding a Scaffold Subunit of the Anaphase-Promoting Complex, Cause Rothmund-Thomson Syndrome Type 1
Abstract
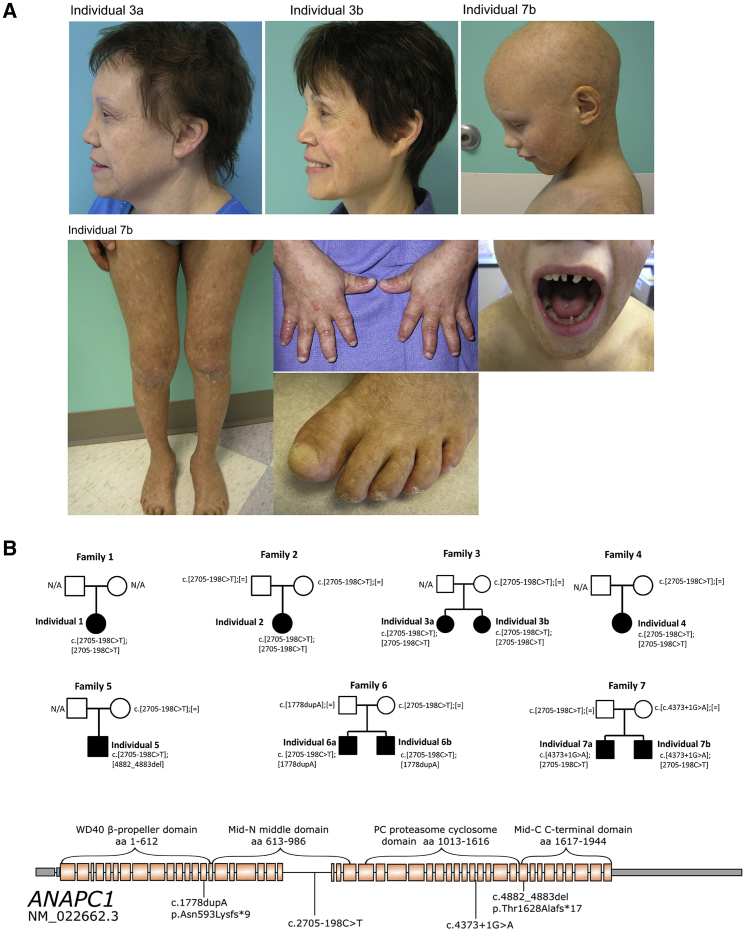
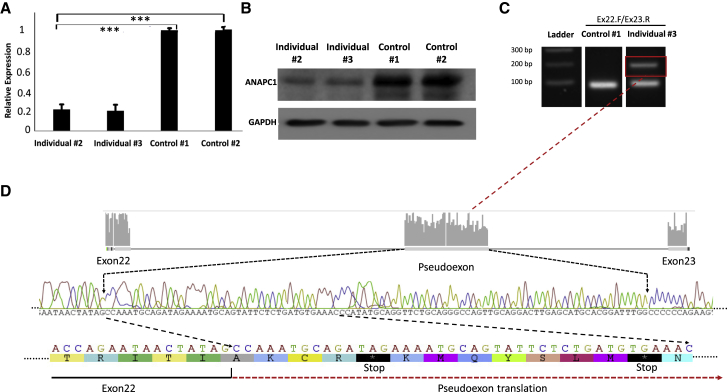
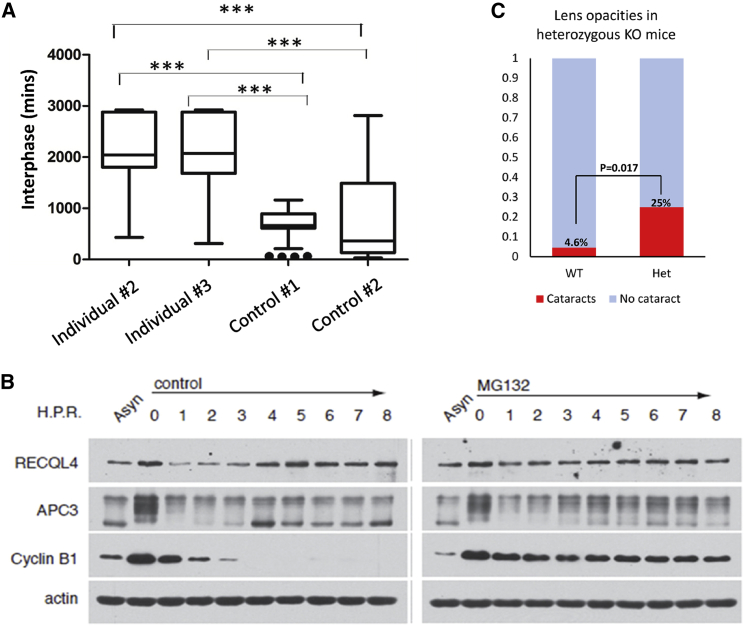
Rothmund-Thomson syndrome (RTS) is an autosomal-recessive disorder characterized by poikiloderma, sparse hair, short stature, and skeletal anomalies. Type 2 RTS, which is defined by the presence of bi-allelic mutations in RECQL4, is characterized by increased cancer susceptibility and skeletal anomalies, whereas the genetic basis of RTS type 1, which is associated with juvenile cataracts, is unknown. We studied ten individuals, from seven families, who had RTS type 1 and identified a deep intronic splicing mutation of the ANAPC1 gene, a component of the anaphase-promoting complex/cyclosome (APC/C), in all affected individuals, either in the homozygous state or in trans with another mutation. Fibroblast studies showed that the intronic mutation causes the activation of a 95 bp pseudoexon, leading to mRNAs with premature termination codons and nonsense-mediated decay, decreased ANAPC1 protein levels, and prolongation of interphase. Interestingly, mice that were heterozygous for a knockout mutation have an increased incidence of cataracts. Our results demonstrate that deficiency in the APC/C is a cause of RTS type 1 and suggest a possible link between the APC/C and RECQL4 helicase because both proteins are involved in DNA repair and replication.
Keywords: ANAPC1; RECQL4; Rothmund-Thomson syndrome; alternative splicing; anaphase-promoting complex; cataracts; cryptic splice site; poikiloderma; pseudoexon; splicing variant.
Copyright © 2019 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Wang L.L., Plon S.E. Rothmund-Thomson syndrome. In: Adam M.P., Ardinger H.H., Pagon R.A., Wallace S.E., Bean L.J.H., Stephens K., Amemiya A., editors. In GeneReviews. Seattle; 1993.
-
- Mehollin-Ray A.R., Kozinetz C.A., Schlesinger A.E., Guillerman R.P., Wang L.L. Radiographic abnormalities in Rothmund-Thomson syndrome and genotype-phenotype correlation with RECQL4 mutation status. AJR Am. J. Roentgenol. 2008;191 W62-6. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
