

De Novo Variants Disturbing the Transactivation Capacity of POU3F3 Cause a Characteristic Neurodevelopmental Disorder
- PMID: 31303265
- PMCID: PMC6698880
- DOI: 10.1016/j.ajhg.2019.06.007
De Novo Variants Disturbing the Transactivation Capacity of POU3F3 Cause a Characteristic Neurodevelopmental Disorder
Abstract
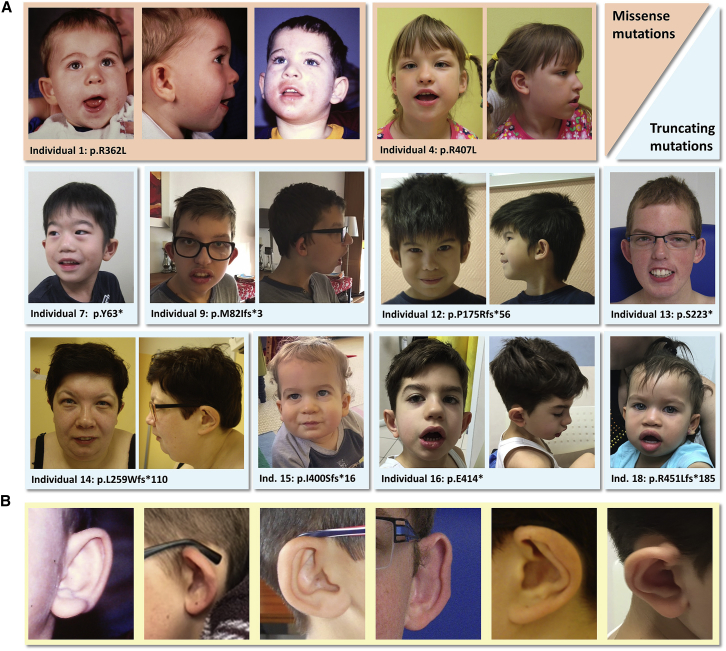
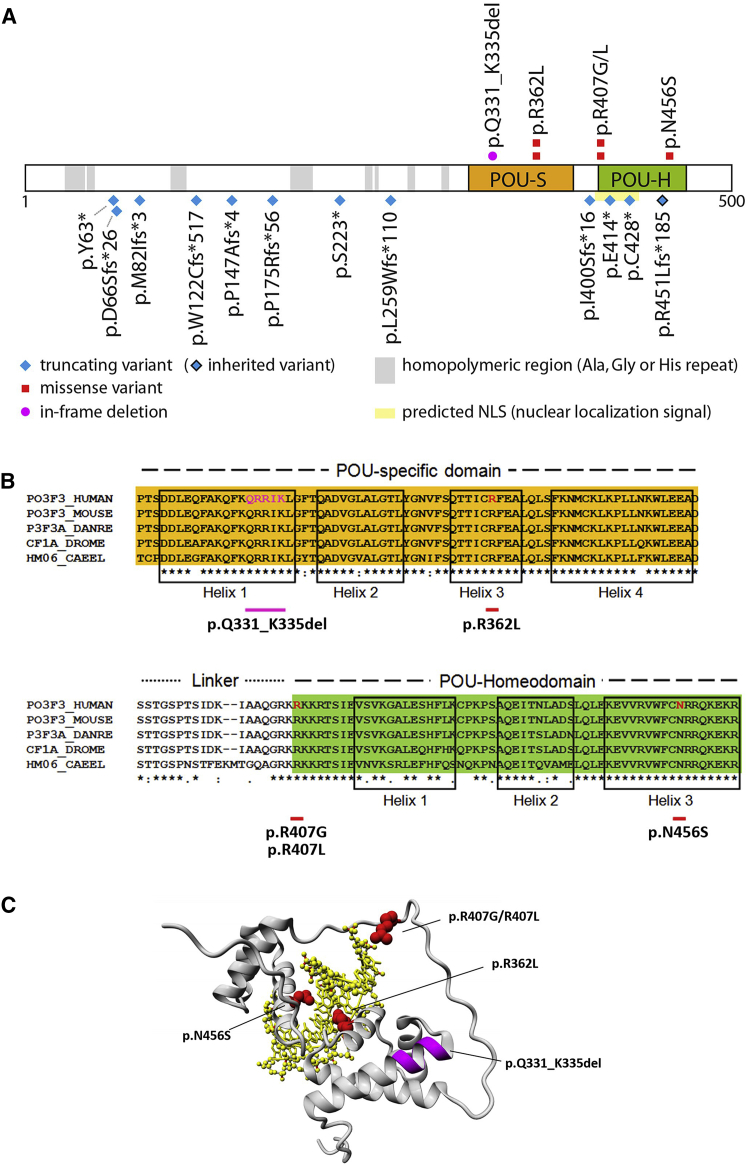
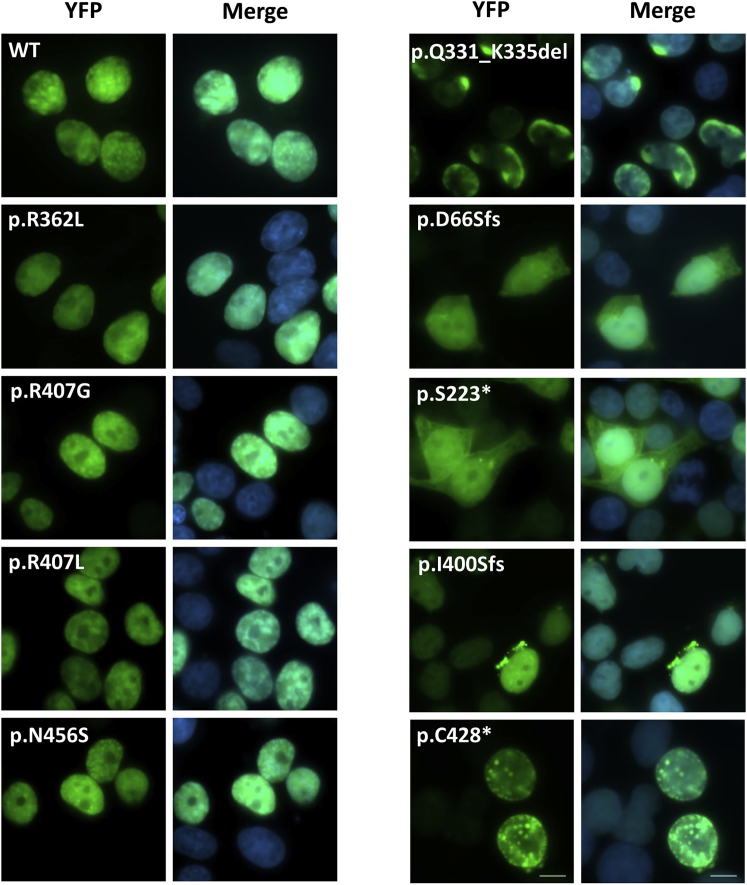
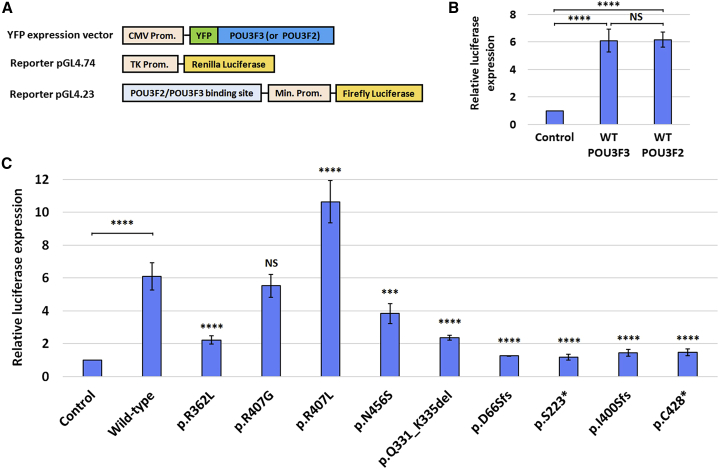
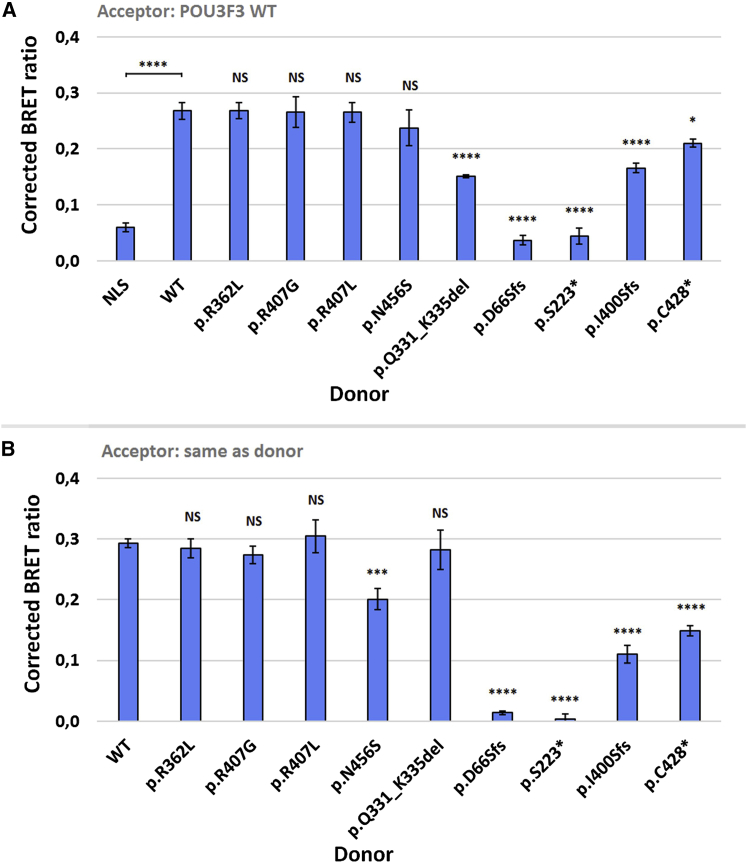
POU3F3, also referred to as Brain-1, is a well-known transcription factor involved in the development of the central nervous system, but it has not previously been associated with a neurodevelopmental disorder. Here, we report the identification of 19 individuals with heterozygous POU3F3 disruptions, most of which are de novo variants. All individuals had developmental delays and/or intellectual disability and impairments in speech and language skills. Thirteen individuals had characteristic low-set, prominent, and/or cupped ears. Brain abnormalities were observed in seven of eleven MRI reports. POU3F3 is an intronless gene, insensitive to nonsense-mediated decay, and 13 individuals carried protein-truncating variants. All truncating variants that we tested in cellular models led to aberrant subcellular localization of the encoded protein. Luciferase assays demonstrated negative effects of these alleles on transcriptional activation of a reporter with a FOXP2-derived binding motif. In addition to the loss-of-function variants, five individuals had missense variants that clustered at specific positions within the functional domains, and one small in-frame deletion was identified. Two missense variants showed reduced transactivation capacity in our assays, whereas one variant displayed gain-of-function effects, suggesting a distinct pathophysiological mechanism. In bioluminescence resonance energy transfer (BRET) interaction assays, all the truncated POU3F3 versions that we tested had significantly impaired dimerization capacities, whereas all missense variants showed unaffected dimerization with wild-type POU3F3. Taken together, our identification and functional cell-based analyses of pathogenic variants in POU3F3, coupled with a clinical characterization, implicate disruptions of this gene in a characteristic neurodevelopmental disorder.
Keywords: BRET assay; Brain-1; FOXP2; POU3F2; POU3F3; de novo variants; intellectual disability; luciferase reporter; speech/language disorder.
Copyright © 2019 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Z.P., S.T., and D.N.S. are full-time employees of Ambry Genetics. Exome sequencing is one of Ambry’s commercially available tests. The authors declare no other competing interests.
Figures
References
-
- Sumiyama K., Washio-Watanabe K., Saitou N., Hayakawa T., Ueda S. Class III POU genes: Generation of homopolymeric amino acid repeats under GC pressure in mammals. J. Mol. Evol. 1996;43:170–178. - PubMed
-
- He X., Treacy M.N., Simmons D.M., Ingraham H.A., Swanson L.W., Rosenfeld M.G. Expression of a large family of POU-domain regulatory genes in mammalian brain development. Nature. 1989;340:35–41. - PubMed
-
- Hagino-Yamagishi K., Saijoh Y., Ikeda M., Ichikawa M., Minamikawa-Tachino R., Hamada H. Predominant expression of Brn-2 in the postmitotic neurons of the developing mouse neocortex. Brain Res. 1997;752:261–268. - PubMed
-
- McEvilly R.J., de Diaz M.O., Schonemann M.D., Hooshmand F., Rosenfeld M.G. Transcriptional regulation of cortical neuron migration by POU domain factors. Science. 2002;295:1528–1532. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
