

The bread wheat epigenomic map reveals distinct chromatin architectural and evolutionary features of functional genetic elements
- PMID: 31307500
- PMCID: PMC6628505
- DOI: 10.1186/s13059-019-1746-8
The bread wheat epigenomic map reveals distinct chromatin architectural and evolutionary features of functional genetic elements
Abstract
Background: Bread wheat is an allohexaploid species with a 16-Gb genome that has large intergenic regions, which presents a big challenge for pinpointing regulatory elements and further revealing the transcriptional regulatory mechanisms. Chromatin profiling to characterize the combinatorial patterns of chromatin signatures is a powerful means to detect functional elements and clarify regulatory activities in human studies.
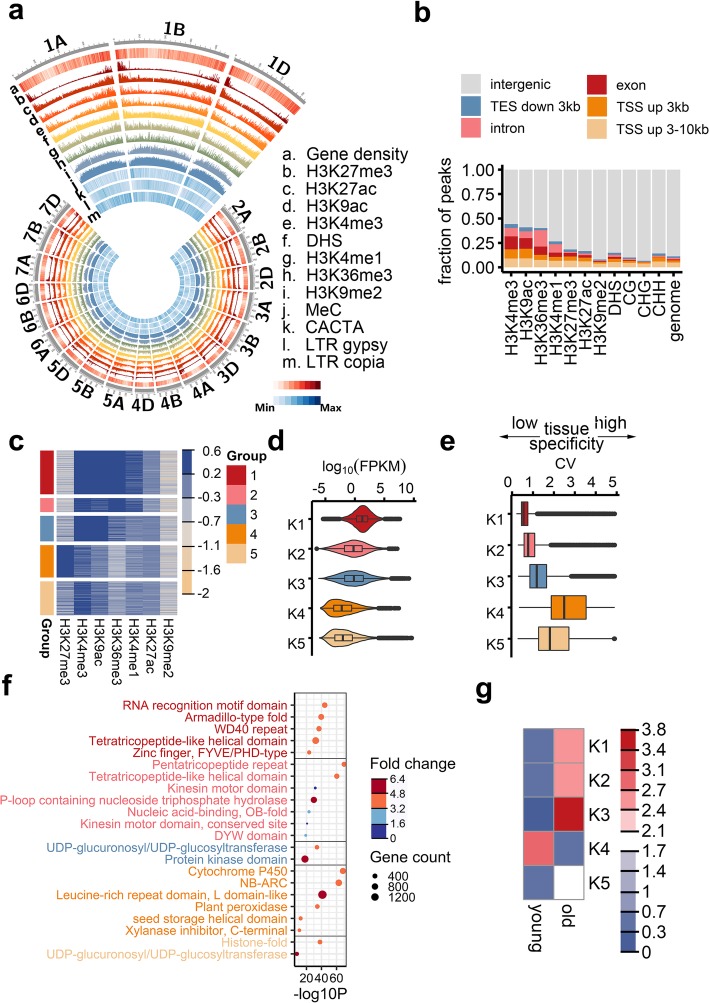
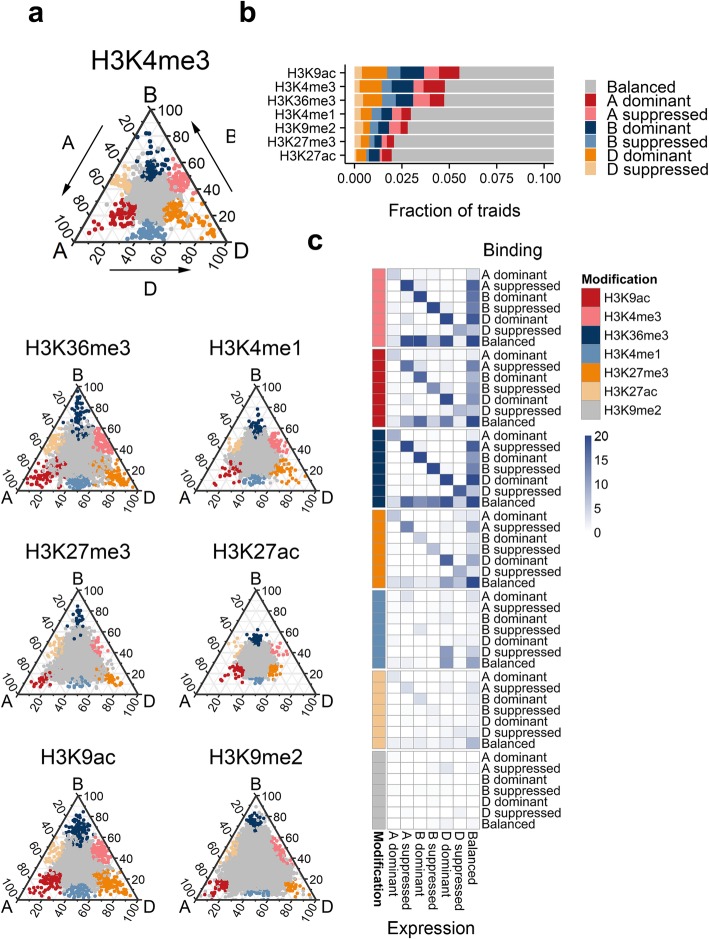
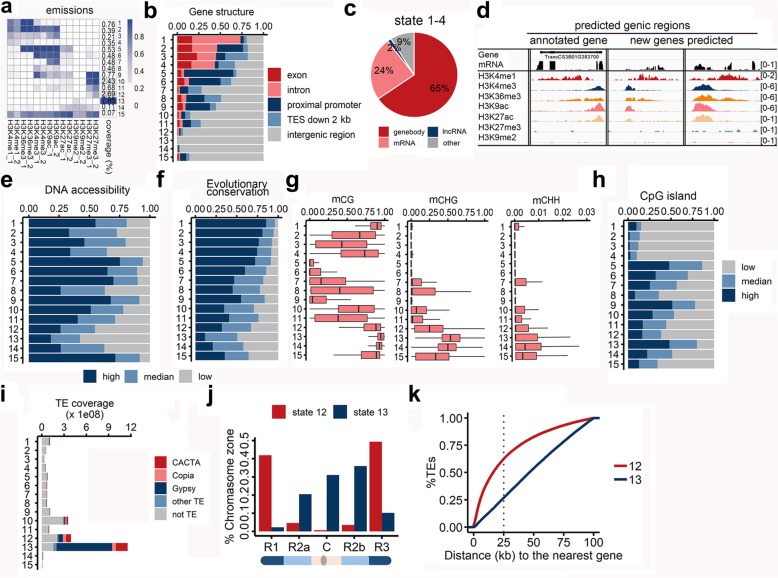
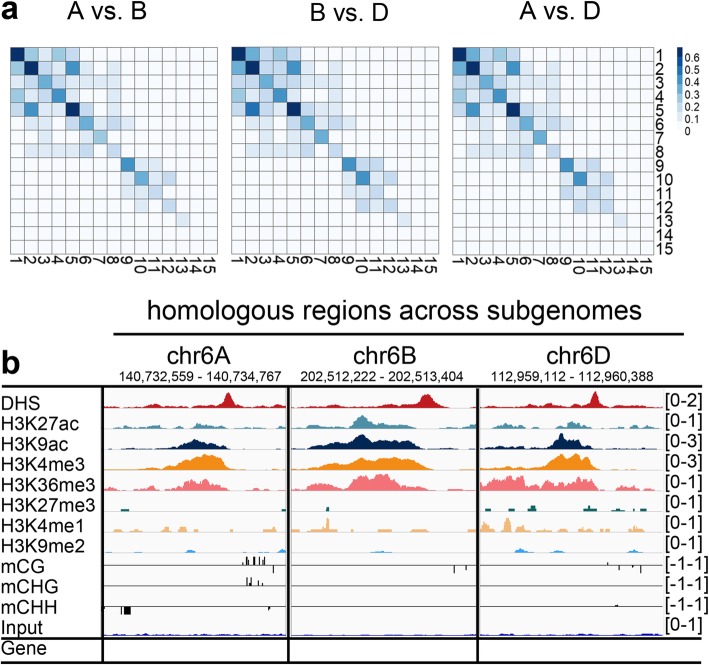
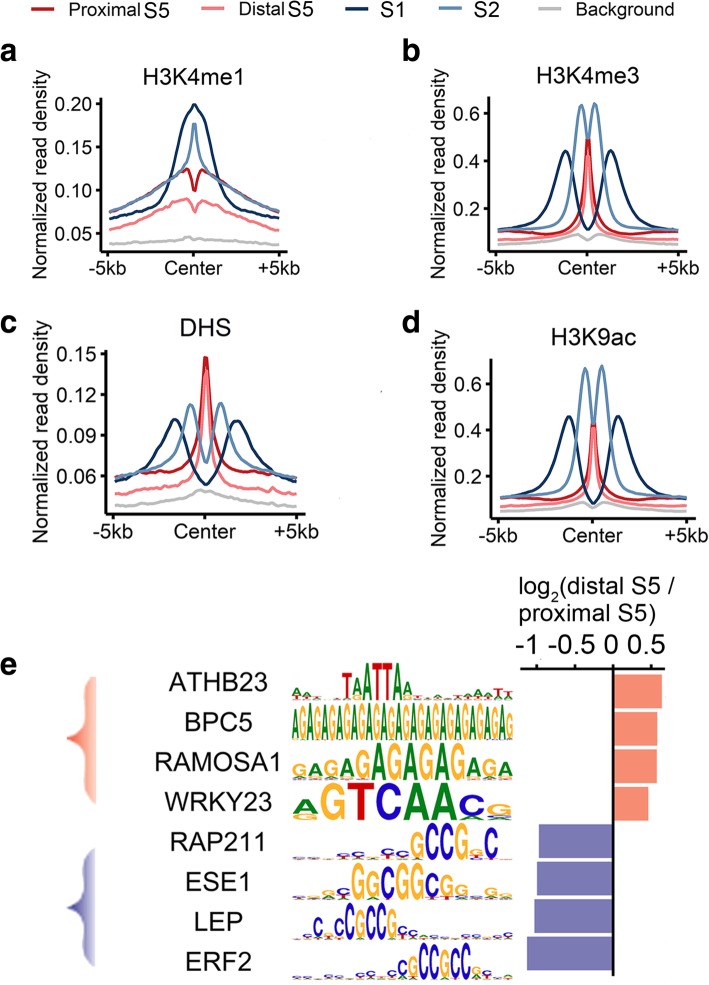
Results: In the present study, through comprehensive analyses of the open chromatin, DNA methylome, seven major chromatin marks, and transcriptomic data generated for seedlings of allohexaploid wheat, we detected distinct chromatin architectural features surrounding various functional elements, including genes, promoters, enhancer-like elements, and transposons. Thousands of new genic regions and cis-regulatory elements are identified based on the combinatorial pattern of chromatin features. Roughly 1.5% of the genome encodes a subset of active regulatory elements, including promoters and enhancer-like elements, which are characterized by a high degree of chromatin openness and histone acetylation, an abundance of CpG islands, and low DNA methylation levels. A comparison across sub-genomes reveals that evolutionary selection on gene regulation is targeted at the sequence and chromatin feature levels. The divergent enrichment of cis-elements between enhancer-like sequences and promoters implies these functional elements are targeted by different transcription factors.
Conclusions: We herein present a systematic epigenomic map for the annotation of cis-regulatory elements in the bread wheat genome, which provides new insights into the connections between chromatin modifications and cis-regulatory activities in allohexaploid wheat.
Keywords: Allohexaploid; Bread wheat; Chromatin signature; Enhancer; Epigenomic map; Promoter; Regulatory element.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures

References
-
- International Wheat Genome Sequencing C, investigators IRp, Appels R, Eversole K, Feuillet C, Keller B, Rogers J, Stein N, investigators Iw-gap, Pozniak CJ, et al. Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science. 2018;361:eaar7191. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
