

Targeting MUC1-C Inhibits TWIST1 Signaling in Triple-Negative Breast Cancer
- PMID: 31308076
- PMCID: PMC6774902
- DOI: 10.1158/1535-7163.MCT-19-0156
Targeting MUC1-C Inhibits TWIST1 Signaling in Triple-Negative Breast Cancer
Abstract
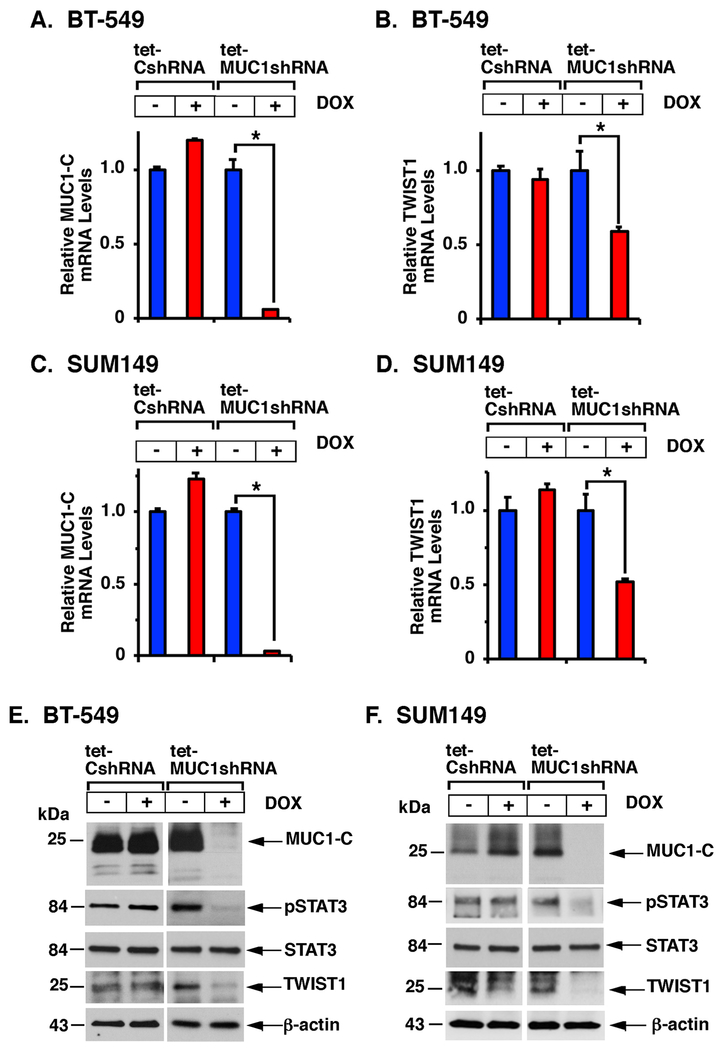
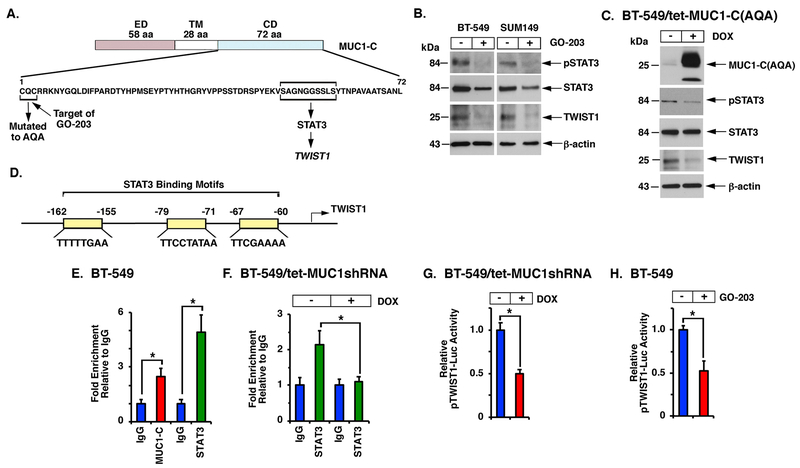
The oncogenic MUC1-C protein and the TWIST1 epithelial-mesenchymal transition transcription factor (EMT-TF) are aberrantly expressed in triple-negative breast cancer (TNBC) cells. However, there is no known association between MUC1-C and TWIST1 in TNBC or other cancer cells. Here, we show that MUC1-C activates STAT3, and that MUC1-C and pSTAT3 drive induction of the TWIST1 gene. In turn, MUC1-C binds directly to TWIST1, and MUC1-C/TWIST1 complexes activate MUC1-C expression in an autoinductive circuit. The functional significance of the MUC1-C/TWIST1 circuit is supported by the demonstration that this pathway is sufficient for driving (i) the EMT-TFs, ZEB1 and SNAIL, (ii) multiple genes in the EMT program as determined by RNA-seq, and (iii) the capacity for cell invasion. We also demonstrate that the MUC1-C/TWIST1 circuit drives (i) expression of the stem cell markers SOX2, BMI1, ALDH1, and CD44, (ii) self-renewal capacity, and (iii) tumorigenicity. In concert with these results, we show that MUC1-C and TWIST1 also drive EMT and stemness in association with acquired paclitaxel (PTX) resistance. Of potential therapeutic importance, targeting MUC1-C and thereby TWIST1 reverses the PTX refractory phenotype as evidenced by synergistic activity with PTX against drug-resistant cells. These findings uncover a master role for MUC1-C in driving the induction of TWIST1, EMT, stemness, and drug resistance, and support MUC1-C as a highly attractive target for inhibiting TNBC plasticity and progression.
©2019 American Association for Cancer Research.
Conflict of interest statement
Figures




Similar articles
-
Modulation of SOX2 expression delineates an end-point for paclitaxel-effectiveness in breast cancer stem cells.Sci Rep. 2017 Aug 23;7(1):9170. doi: 10.1038/s41598-017-08971-2. Sci Rep. 2017. PMID: 28835684 Free PMC article.
-
α-Linolenic acid inhibits the migration of human triple-negative breast cancer cells by attenuating Twist1 expression and suppressing Twist1-mediated epithelial-mesenchymal transition.Biochem Pharmacol. 2020 Oct;180:114152. doi: 10.1016/j.bcp.2020.114152. Epub 2020 Jul 15. Biochem Pharmacol. 2020. PMID: 32679125
-
LINC01638 lncRNA activates MTDH-Twist1 signaling by preventing SPOP-mediated c-Myc degradation in triple-negative breast cancer.Oncogene. 2018 Nov;37(47):6166-6179. doi: 10.1038/s41388-018-0396-8. Epub 2018 Jul 12. Oncogene. 2018. PMID: 30002443
-
Epithelial-mesenchymal transition and cancer stemness: the Twist1-Bmi1 connection.Biosci Rep. 2011 Dec;31(6):449-55. doi: 10.1042/BSR20100114. Biosci Rep. 2011. PMID: 21919891 Review.
-
The role of TWIST1 in epithelial-mesenchymal transition and cancers.Tumour Biol. 2016 Jan;37(1):185-97. doi: 10.1007/s13277-015-4450-7. Epub 2015 Nov 24. Tumour Biol. 2016. PMID: 26602382 Review.
Cited by
-
Addiction of Merkel cell carcinoma to MUC1-C identifies a potential new target for treatment.Oncogene. 2022 Jul;41(27):3511-3523. doi: 10.1038/s41388-022-02361-3. Epub 2022 Jun 10. Oncogene. 2022. PMID: 35688945 Free PMC article.
-
High-Throughput Screen of Natural Compounds and Biomarkers for NSCLC Treatment by Differential Expression and Weighted Gene Coexpression Network Analysis (WGCNA).Biomed Res Int. 2021 Aug 26;2021:5955343. doi: 10.1155/2021/5955343. eCollection 2021. Biomed Res Int. 2021. Retraction in: Biomed Res Int. 2024 Mar 20;2024:9892473. doi: 10.1155/2024/9892473. PMID: 34485520 Free PMC article. Retracted.
-
Patterns of immune evasion in triple-negative breast cancer and new potential therapeutic targets: a review.Front Immunol. 2024 Dec 13;15:1513421. doi: 10.3389/fimmu.2024.1513421. eCollection 2024. Front Immunol. 2024. PMID: 39735530 Free PMC article. Review.
-
The Significance of Aldehyde Dehydrogenase 1 in Cancers.Int J Mol Sci. 2024 Dec 30;26(1):251. doi: 10.3390/ijms26010251. Int J Mol Sci. 2024. PMID: 39796106 Free PMC article. Review.
-
The Oncoprotein Mucin 1 in Pancreatic Cancer Onset and Progression: Potential Clinical Implications.Biomolecules. 2025 Feb 13;15(2):275. doi: 10.3390/biom15020275. Biomolecules. 2025. PMID: 40001578 Free PMC article. Review.
References
-
- Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer 2002;2(6):442–54. - PubMed
-
- Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell 2009;139(5):871–90. - PubMed
-
- Li R, Liang J, Ni S, Zhou T, Qing X, Li H, et al. A mesenchymal-to-epithelial transition initiates and is required for the nuclear reprogramming of mouse fibroblasts. Cell Stem Cell 2010;7(1):51–63. - PubMed
-
- De Craene B, Berx G. Regulatory networks defining EMT during cancer initiation and progression. Nat Rev Cancer 2013;13(2):97–110. - PubMed
-
- Skrypek N, Goossens S, De Smedt E, Vandamme N, Berx G. Epithelial-to-mesenchymal transition: epigenetic reprogramming driving cellular plasticity. Trends Genet 2017;33(12):943–59. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
