

De novo X-linked Alport syndrome in a 3-year-old girl
- PMID: 31308192
- PMCID: PMC6663154
- DOI: 10.1136/bcr-2019-230183
De novo X-linked Alport syndrome in a 3-year-old girl
Abstract
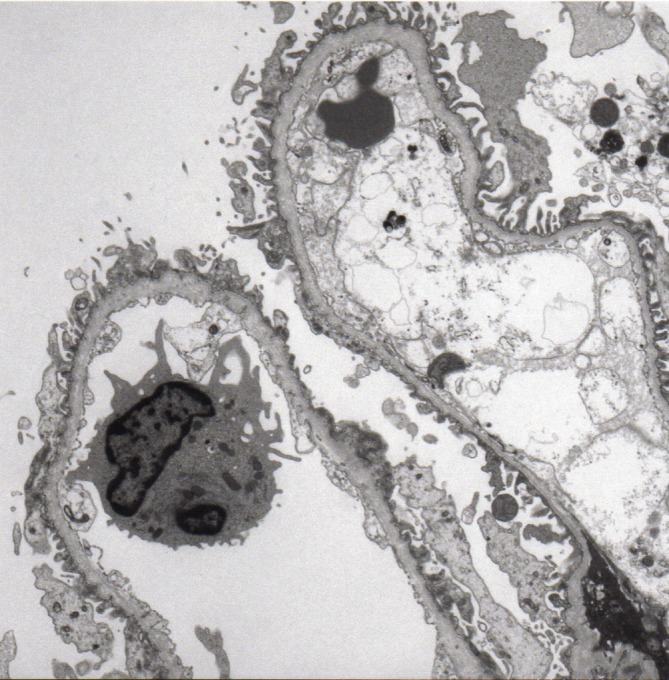
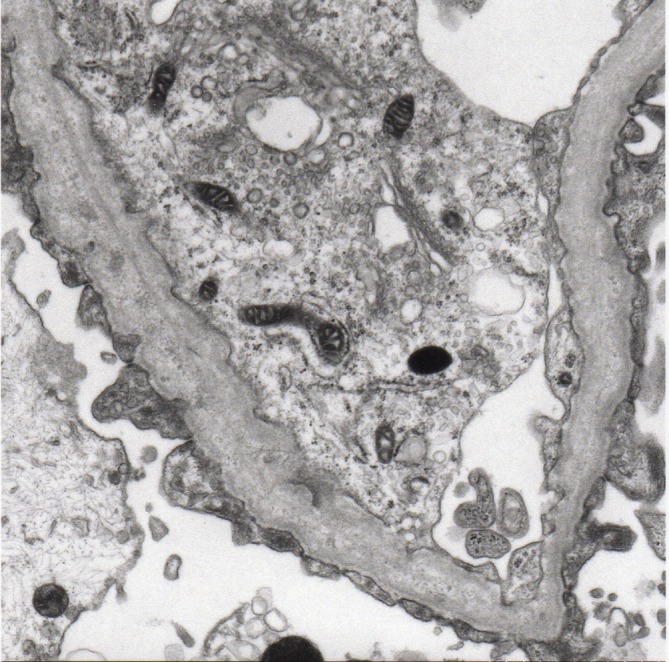
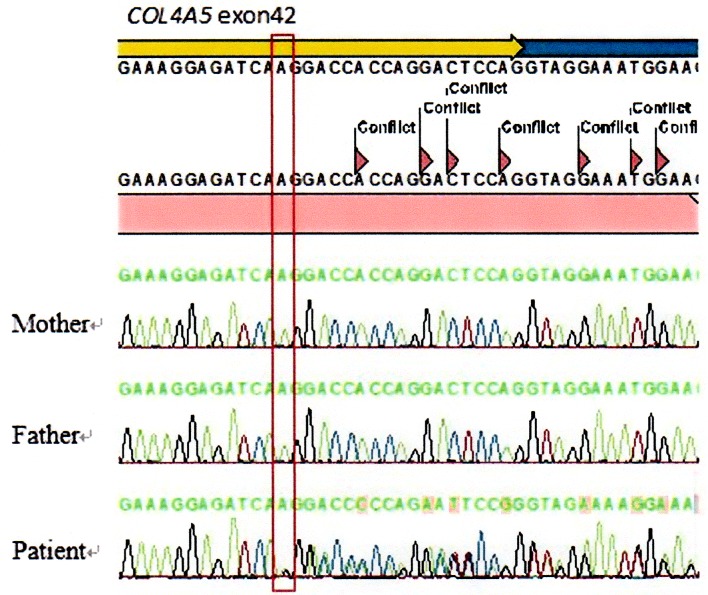
Alport syndrome (AS) is an inherited kidney disease that may lead to end-stage renal disease in early adult life. It is a clinically and genetically heterogeneous nephropathy. The possibility of a patient with haematuria or proteinuria being diagnosed as having AS cannot be excluded even if the patient is female or if the family history is unknown. We report a 3-year-old girl with a de novo frameshift mutation, c.3906delA p.(Gly1303Aspfs*17), in the COL4A5 gene. The significance of the electron microscopic study on the glomerular basement membrane must be emphasised because it is the first step towards the diagnosis of AS. Genetic analysis provides the only conclusive diagnosis of AS, by determining the mode of inheritance and prognosis.
Keywords: genetic screening counselling; proteinurea; urinary and genital tract disorders.
© BMJ Publishing Group Limited 2019. No commercial re-use. See rights and permissions. Published by BMJ.
Conflict of interest statement
Competing interests: None declared.
Figures
Similar articles
-
Alport syndrome: deducing the mode of inheritance from the presence of haematuria in family members.Pediatr Nephrol. 2020 Jan;35(1):59-66. doi: 10.1007/s00467-018-4121-1. Epub 2018 Nov 30. Pediatr Nephrol. 2020. PMID: 30506145 Review.
-
Autosomal dominant Alport syndrome: molecular analysis of the COL4A4 gene and clinical outcome.Nephrol Dial Transplant. 2009 May;24(5):1464-71. doi: 10.1093/ndt/gfn681. Epub 2009 Jan 7. Nephrol Dial Transplant. 2009. PMID: 19129241
-
[Collagen type IV nephropathy: from thin basement membrane nephropathy to Alport syndrome].Orv Hetil. 2005 Dec 25;146(52):2647-53. Orv Hetil. 2005. PMID: 16468607 Hungarian.
-
A novel frameshift mutation of COL4A5 in a Chinese family with presumed IgA nephropathy and chronic glomerulonephritis.J Clin Lab Anal. 2020 Dec;34(12):e23558. doi: 10.1002/jcla.23558. Epub 2020 Sep 6. J Clin Lab Anal. 2020. PMID: 32893410 Free PMC article.
-
Alport syndrome and thin glomerular basement membrane nephropathy: a practical approach to diagnosis.Arch Pathol Lab Med. 2009 Feb;133(2):224-32. doi: 10.5858/133.2.224. Arch Pathol Lab Med. 2009. PMID: 19195966 Review.
Cited by
-
Preimplantation Genetic Testing Prevented Intergenerational Transmission of X-Linked Alport Syndrome.Kidney Dis (Basel). 2021 Sep 9;7(6):514-520. doi: 10.1159/000517796. eCollection 2021 Nov. Kidney Dis (Basel). 2021. PMID: 34901197 Free PMC article.
References
-
- Jais JP, Knebelmann B, Giatras I, et al. . X-linked Alport syndrome: natural history and genotype-phenotype correlations in girls and women belonging to 195 families: a "European Community Alport Syndrome Concerted Action" study. J Am Soc Nephrol 2003;14:2603–10. 10.1097/01.ASN.0000090034.71205.74 - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials