

Pleiotropic effects for Parkin and LRRK2 in leprosy type-1 reactions and Parkinson's disease
- PMID: 31308240
- PMCID: PMC6681704
- DOI: 10.1073/pnas.1901805116
Pleiotropic effects for Parkin and LRRK2 in leprosy type-1 reactions and Parkinson's disease
Abstract
Type-1 reactions (T1R) are pathological inflammatory episodes and main contributors to nerve damage in leprosy. Here, we evaluate the genewise enrichment of rare protein-altering variants in 7 genes where common variants were previously associated with T1R. We selected 474 Vietnamese leprosy patients of which 237 were T1R-affected and 237 were T1R-free matched controls. Genewise enrichment of nonsynonymous variants was tested with both kernel-based (sequence kernel association test [SKAT]) and burden methods. Of the 7 genes tested 2 showed statistical evidence of association with T1R. For the LRRK2 gene an enrichment of nonsynonymous variants was observed in T1R-free controls (PSKAT-O = 1.6 × 10-4). This genewise association was driven almost entirely by the gain-of-function variant R1628P (P = 0.004; odds ratio = 0.29). The second genewise association was found for the Parkin coding gene PRKN (formerly PARK2) where 7 rare variants were enriched in T1R-affected cases (PSKAT-O = 7.4 × 10-5). Mutations in both PRKN and LRRK2 are known causes of Parkinson's disease (PD). Hence, we evaluated to what extent such rare amino acid changes observed in T1R are shared with PD. We observed that amino acids in Parkin targeted by nonsynonymous T1R-risk mutations were also enriched for mutations implicated in PD (P = 1.5 × 10-4). Hence, neuroinflammation in PD and peripheral nerve damage due to inflammation in T1R share overlapping genetic control of pathogenicity.
Keywords: LRRK2; Parkin; Parkinson’s disease; inflammation; leprosy type-1 reaction.
Copyright © 2019 the Author(s). Published by PNAS.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

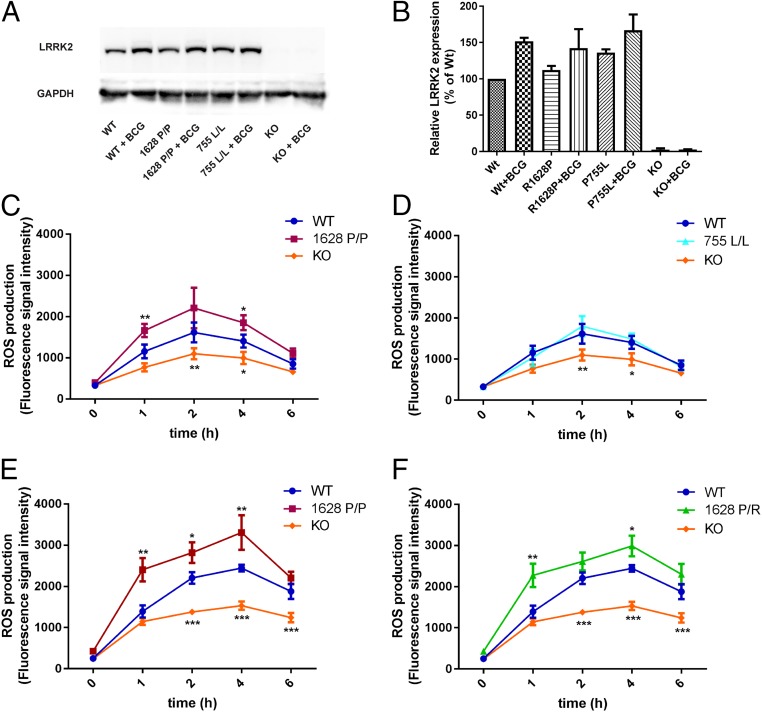

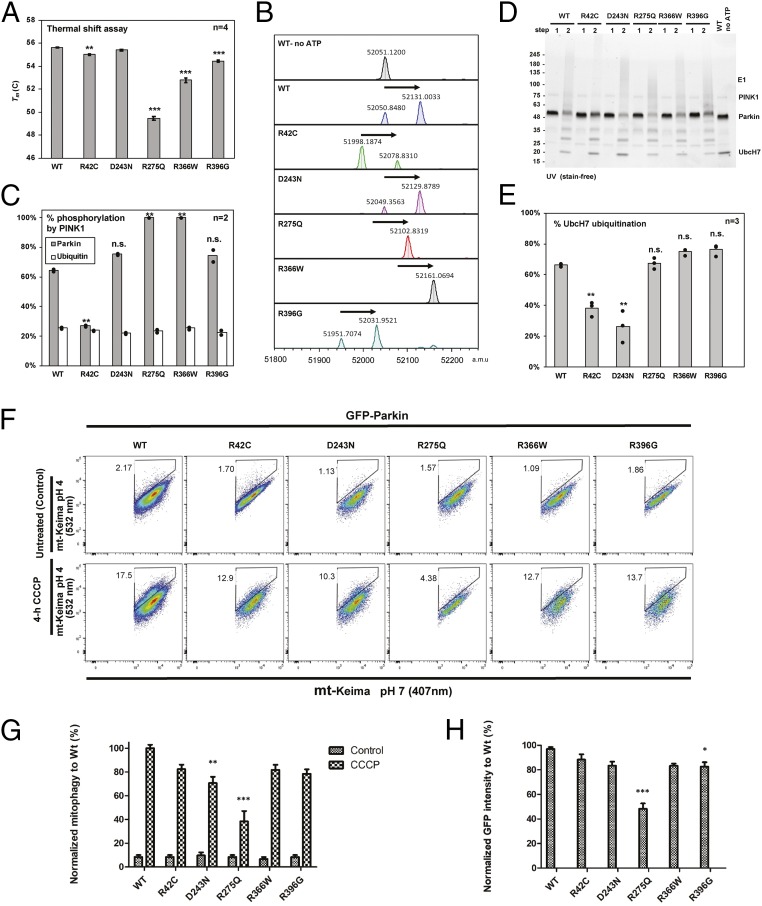
Comment in
-
Is there an antagonistic pleiotropic effect of a LRRK2 mutation on leprosy and Parkinson's disease?Proc Natl Acad Sci U S A. 2020 May 12;117(19):10122-10123. doi: 10.1073/pnas.2000533117. Epub 2020 Apr 28. Proc Natl Acad Sci U S A. 2020. PMID: 32345713 Free PMC article. No abstract available.
-
Reply to Zhang et al.: The differential role of LRRK2 variants in nested leprosy phenotypes.Proc Natl Acad Sci U S A. 2020 May 12;117(19):10124-10125. doi: 10.1073/pnas.2002654117. Epub 2020 Apr 28. Proc Natl Acad Sci U S A. 2020. PMID: 32345724 Free PMC article. No abstract available.
Similar articles
-
A Missense LRRK2 Variant Is a Risk Factor for Excessive Inflammatory Responses in Leprosy.PLoS Negl Trop Dis. 2016 Feb 4;10(2):e0004412. doi: 10.1371/journal.pntd.0004412. eCollection 2016 Feb. PLoS Negl Trop Dis. 2016. PMID: 26844546 Free PMC article.
-
Identification & characterization of leucine-rich repeat kinase 2 & parkin RBR E3 ubiquitin protein ligase variants in patients with Parkinson's disease.Indian J Med Res. 2020 Nov;152(5):498-507. doi: 10.4103/ijmr.IJMR_730_18. Indian J Med Res. 2020. PMID: 33707392 Free PMC article.
-
LRRK2 impairs PINK1/Parkin-dependent mitophagy via its kinase activity: pathologic insights into Parkinson's disease.Hum Mol Genet. 2019 May 15;28(10):1645-1660. doi: 10.1093/hmg/ddz004. Hum Mol Genet. 2019. PMID: 30629163
-
New therapeutic approaches to Parkinson's disease targeting GBA, LRRK2 and Parkin.Neuropharmacology. 2022 Jan 1;202:108822. doi: 10.1016/j.neuropharm.2021.108822. Epub 2021 Oct 7. Neuropharmacology. 2022. PMID: 34626666 Review.
-
Leucine-Rich Repeat Kinase (LRRK2) Genetics and Parkinson's Disease.Adv Neurobiol. 2017;14:3-30. doi: 10.1007/978-3-319-49969-7_1. Adv Neurobiol. 2017. PMID: 28353276 Review.
Cited by
-
Necrosis drives susceptibility to Mycobacterium tuberculosis in PolgD257A mutator mice.bioRxiv [Preprint]. 2024 Dec 2:2024.07.17.603991. doi: 10.1101/2024.07.17.603991. bioRxiv. 2024. Update in: Infect Immun. 2025 Mar 11;93(3):e0032424. doi: 10.1128/iai.00324-24. PMID: 39091776 Free PMC article. Updated. Preprint.
-
Prediction of the occurrence of leprosy reactions based on Bayesian networks.Front Med (Lausanne). 2023 Jul 26;10:1233220. doi: 10.3389/fmed.2023.1233220. eCollection 2023. Front Med (Lausanne). 2023. PMID: 37564037 Free PMC article.
-
Tom20 gates PINK1 activity and mediates its tethering of the TOM and TIM23 translocases upon mitochondrial stress.Proc Natl Acad Sci U S A. 2024 Mar 5;121(10):e2313540121. doi: 10.1073/pnas.2313540121. Epub 2024 Feb 28. Proc Natl Acad Sci U S A. 2024. PMID: 38416681 Free PMC article.
-
Deep resequencing identifies candidate functional genes in leprosy GWAS loci.PLoS Negl Trop Dis. 2021 Dec 8;15(12):e0010029. doi: 10.1371/journal.pntd.0010029. eCollection 2021 Dec. PLoS Negl Trop Dis. 2021. PMID: 34879060 Free PMC article.
-
LRRK2 at the Interface Between Peripheral and Central Immune Function in Parkinson's.Front Neurosci. 2020 May 21;14:443. doi: 10.3389/fnins.2020.00443. eCollection 2020. Front Neurosci. 2020. PMID: 32508566 Free PMC article. Review.
References
-
- Fava V. M., Schurr E., “The complexity of the host genetic contribution to the human response to Mycobacterium leprae” in The International Textbook of Leprosy, Scollard D. M., Gillis T. P., Eds. (American Leprosy Mission, 2016).
-
- Mira M. T., et al. , Susceptibility to leprosy is associated with PARK2 and PACRG. Nature 427, 636–640 (2004). - PubMed
-
- Alcaïs A., et al. , Stepwise replication identifies a low-producing lymphotoxin-alpha allele as a major risk factor for early-onset leprosy. Nat. Genet. 39, 517–522 (2007). - PubMed
-
- Zhang F. R., et al. , Genomewide association study of leprosy. N. Engl. J. Med. 361, 2609–2618 (2009). - PubMed
-
- Zhang F., et al. , Identification of two new loci at IL23R and RAB32 that influence susceptibility to leprosy. Nat. Genet. 43, 1247–1251 (2011). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
