

Full-length RNA profiling reveals pervasive bidirectional transcription terminators in bacteria
- PMID: 31308523
- PMCID: PMC6814526
- DOI: 10.1038/s41564-019-0500-z
Full-length RNA profiling reveals pervasive bidirectional transcription terminators in bacteria
Abstract
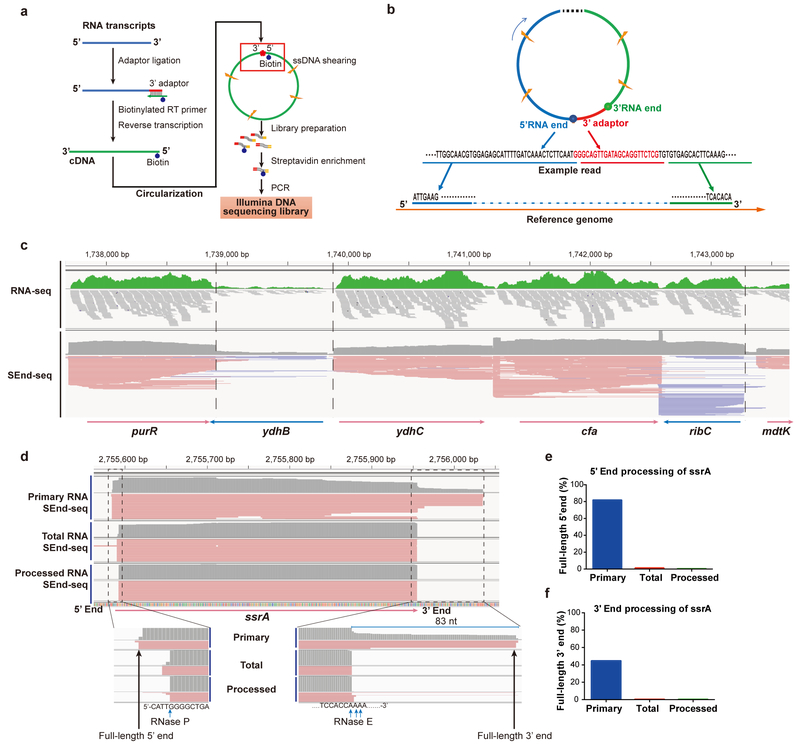
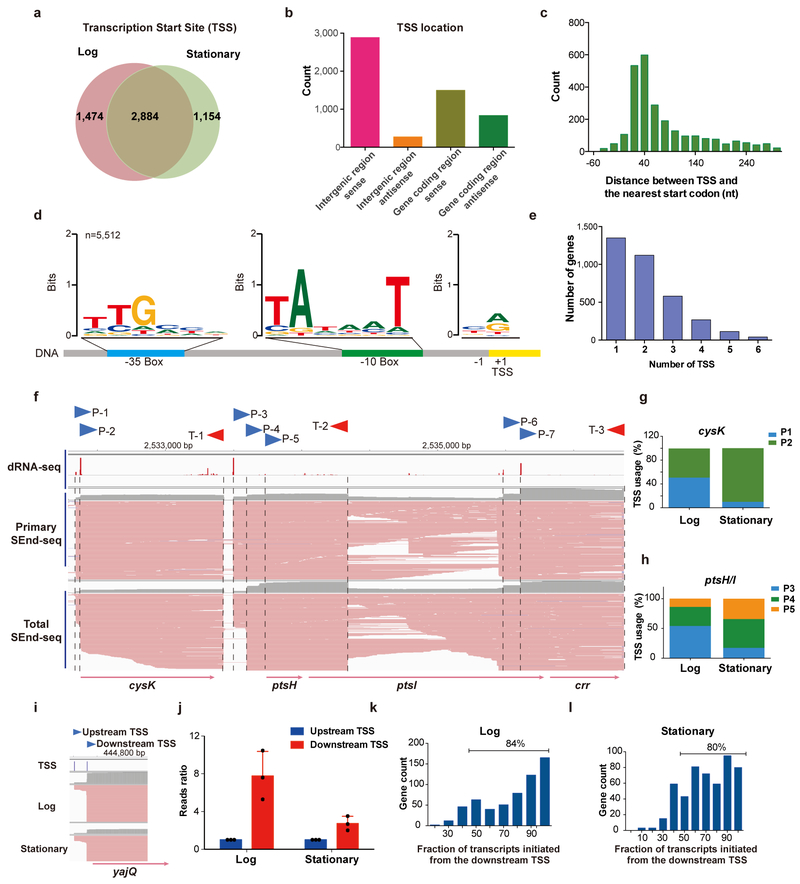
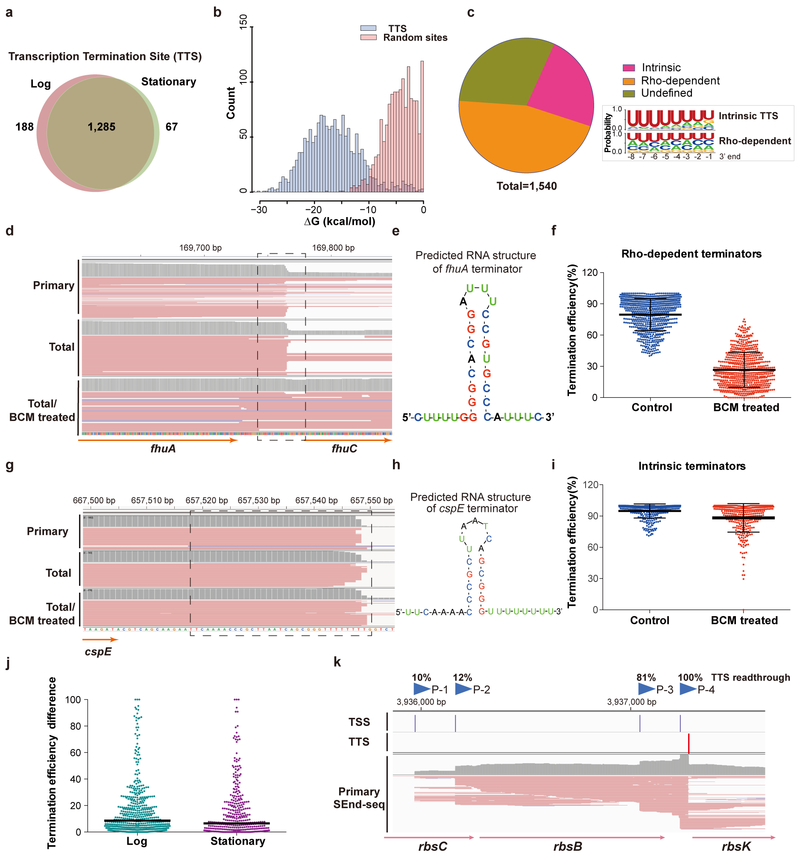
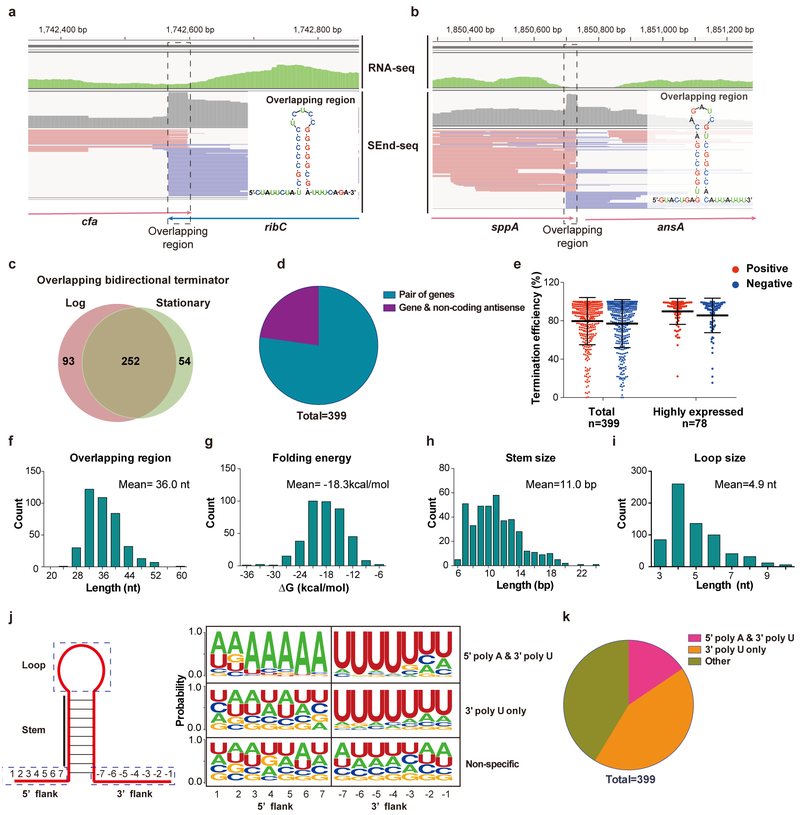
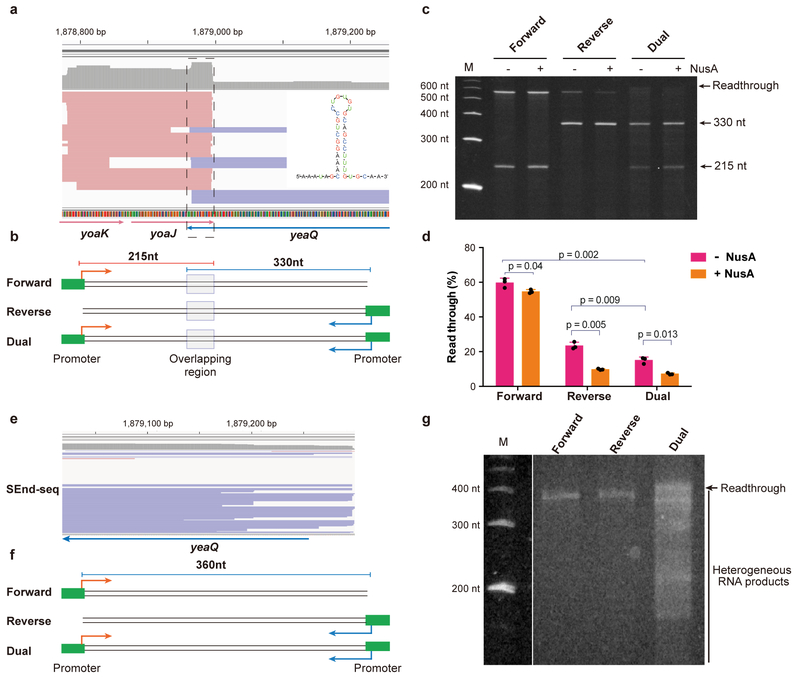
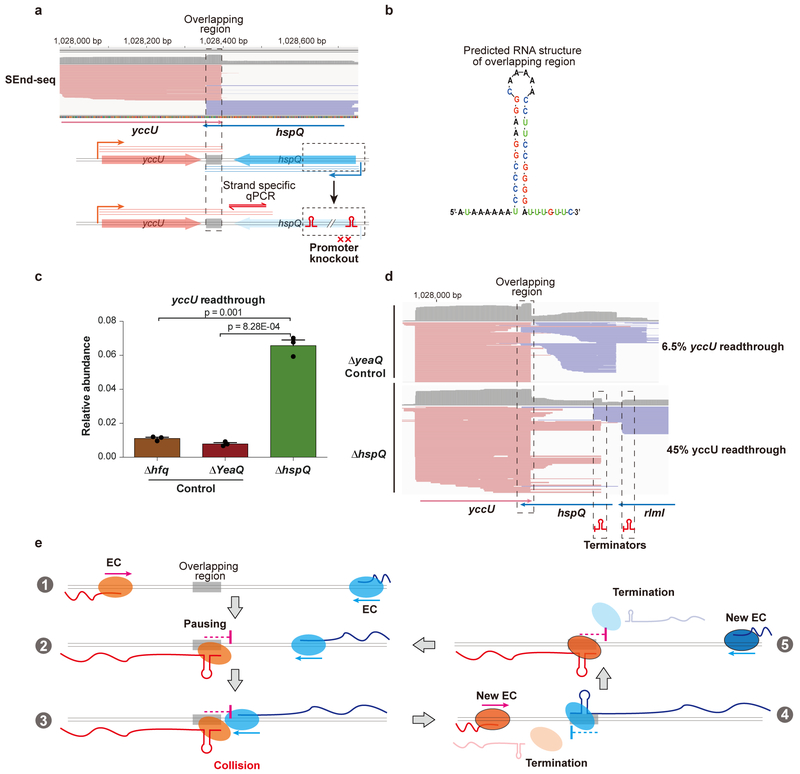
The ability to determine full-length nucleotide composition of individual RNA molecules is essential for understanding the architecture and function of a transcriptome. However, experimental approaches capable of capturing the sequences of both 5' and 3' termini of the same transcript remain scarce. In the present study, simultaneous 5' and 3' end sequencing (SEnd-seq)-a high-throughput and unbiased method that simultaneously maps transcription start and termination sites with single-nucleotide resolution-is presented. Using this method, a comprehensive view of the Escherichia coli transcriptome was obtained, which displays an unexpected level of complexity. SEnd-seq notably expands the catalogue of transcription start sites and termination sites, defines unique transcription units and detects prevalent antisense RNA. Strikingly, the results of the present study unveil widespread overlapping bidirectional terminators located between opposing gene pairs. Furthermore, it has been shown that convergent transcription is a major contributor to highly efficient bidirectional termination both in vitro and in vivo. This finding highlights an underappreciated role of RNA polymerase conflicts in shaping transcript boundaries and suggests an evolutionary strategy for modulating transcriptional output by arranging gene orientation.
Conflict of interest statement
Competing interests
The Rockefeller University has filed a provisional patent application encompassing aspects of the SEnd-seq technology on which S.L. and X.J. are listed as inventors.
Figures
References
-
- Sharma CM et al. The primary transcriptome of the major human pathogen Helicobacter pylori. Nature 464, 250–5 (2010). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
