

Current understanding and treatment of cardiac and skeletal muscle pathology in laminin-α2 chain-deficient congenital muscular dystrophy
- PMID: 31308722
- PMCID: PMC6618038
- DOI: 10.2147/TACG.S187481
Current understanding and treatment of cardiac and skeletal muscle pathology in laminin-α2 chain-deficient congenital muscular dystrophy
Abstract
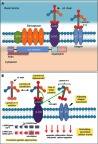
Congenital muscular dystrophy (CMD) is a class of severe early-onset muscular dystrophies affecting skeletal/cardiac muscles as well as the central nervous system (CNS). Laminin-α2 chain-deficient congenital muscular dystrophy (LAMA2 MD), also known as merosin-deficient congenital muscular dystrophy type 1A (MDC1A), is an autosomal recessive CMD characterized by severe muscle weakness and degeneration apparent at birth or in the first 6 months of life. LAMA2 MD is the most common congenital muscular dystrophy, affecting approximately 4 in 500,000 children. The most common cause of death in early-onset LAMA2 MD is respiratory tract infection, with 30% of them dying within the first decade of life. LAMA2 MD is caused by loss-of-function mutations in the LAMA2 gene encoding for the laminin-α2 chain, one of the subunits of laminin-211. Laminin-211 is an extracellular matrix protein that functions to stabilize the basement membrane and muscle fibers during contraction. Since laminin-α2 is expressed in many tissue types including skeletal muscle, cardiac muscle, Schwann cells, and trophoblasts, patients with LAMA2 MD experience a multi-systemic clinical presentation depending on the extent of laminin-α2 chain deficiency. Cardiac manifestations are typically associated with a complete absence of laminin-α2; however, recent case reports highlight cardiac involvement in partial laminin-α2 chain deficiency. Laminin-211 is also expressed in the brain, and many patients have abnormalities on brain imaging; however, mental retardation and/or seizures are rarely seen. Currently, there is no cure for LAMA2 MD, but various therapies are being investigated in an effort to lessen the severity of LAMA2 MD. For example, antisense oligonucleotide-mediated exon skipping and CRISPR-Cas9 genome editing have efficiently restored the laminin-α2 chain in mouse models in vivo. This review consolidates information on the clinical presentation, genetic basis, pathology, and current treatment approaches for LAMA2 MD.
Keywords: CRISPR/Cas9; LAMA2; exon skipping; genome editing; non-homologous end joining; phosphorodiamidate morpholino oligomer.
Conflict of interest statement
The authors report no conflicts of interest in this work.
Figures
References
-
- Quijano-Roy S, Sparks SE, Rutkowski A. LAMA2-Related Muscular Dystrophy. 2012. Jun 7. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2019.
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
