

Functional DNA methylation signatures for autism spectrum disorder genomic risk loci: 16p11.2 deletions and CHD8 variants
- PMID: 31311581
- PMCID: PMC6636171
- DOI: 10.1186/s13148-019-0684-3
Functional DNA methylation signatures for autism spectrum disorder genomic risk loci: 16p11.2 deletions and CHD8 variants
Abstract
Background: Autism spectrum disorder (ASD) is a common and etiologically heterogeneous neurodevelopmental disorder. Although many genetic causes have been identified (> 200 ASD-risk genes), no single gene variant accounts for > 1% of all ASD cases. A role for epigenetic mechanisms in ASD etiology is supported by the fact that many ASD-risk genes function as epigenetic regulators and evidence that epigenetic dysregulation can interrupt normal brain development. Gene-specific DNAm profiles have been shown to assist in the interpretation of variants of unknown significance. Therefore, we investigated the epigenome in patients with ASD or two of the most common genomic variants conferring increased risk for ASD. Genome-wide DNA methylation (DNAm) was assessed using the Illumina Infinium HumanMethylation450 and MethylationEPIC arrays in blood from individuals with ASD of heterogeneous, undefined etiology (n = 52), and individuals with 16p11.2 deletions (16p11.2del, n = 9) or pathogenic variants in the chromatin modifier CHD8 (CHD8+/-, n = 7).
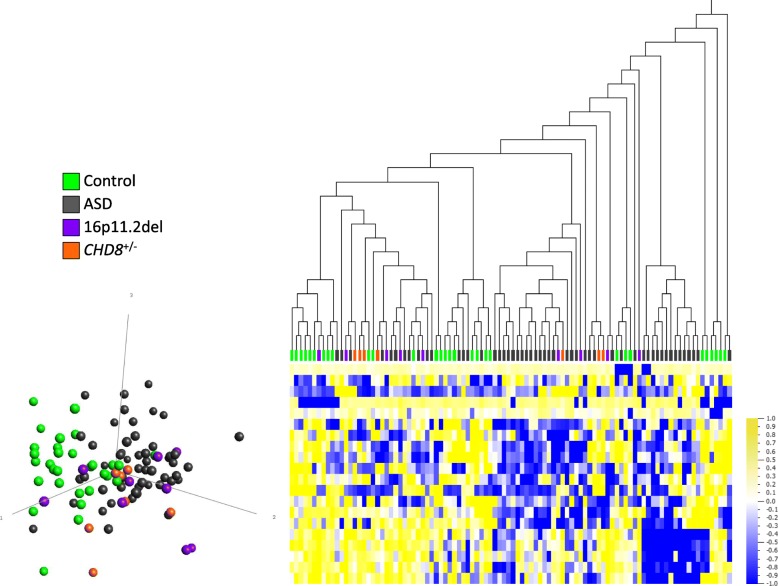
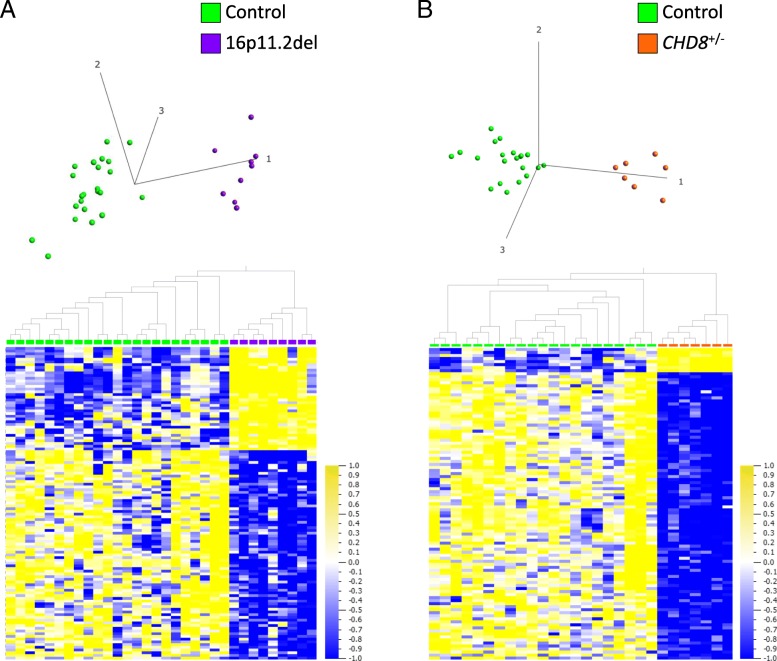
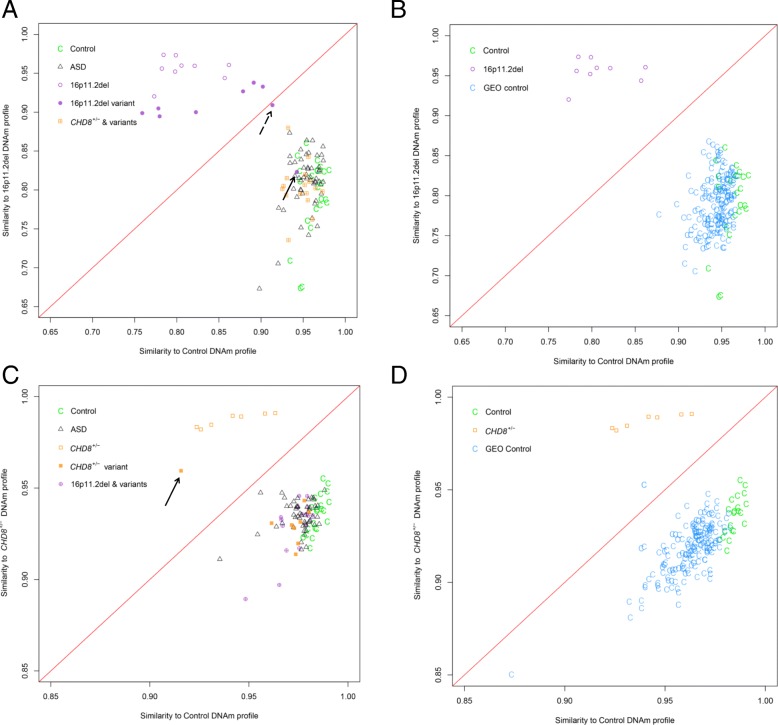
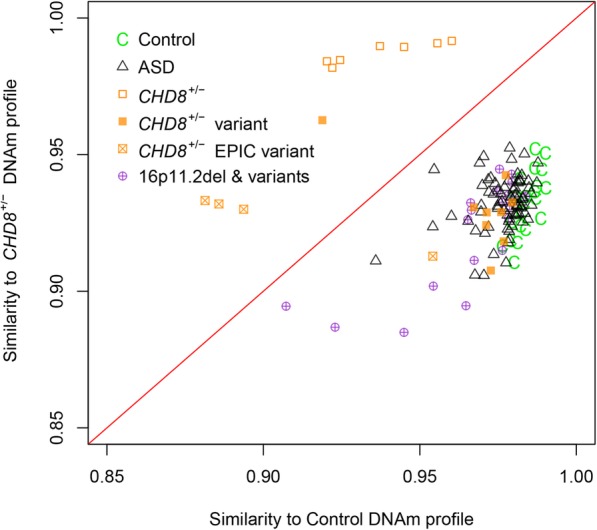
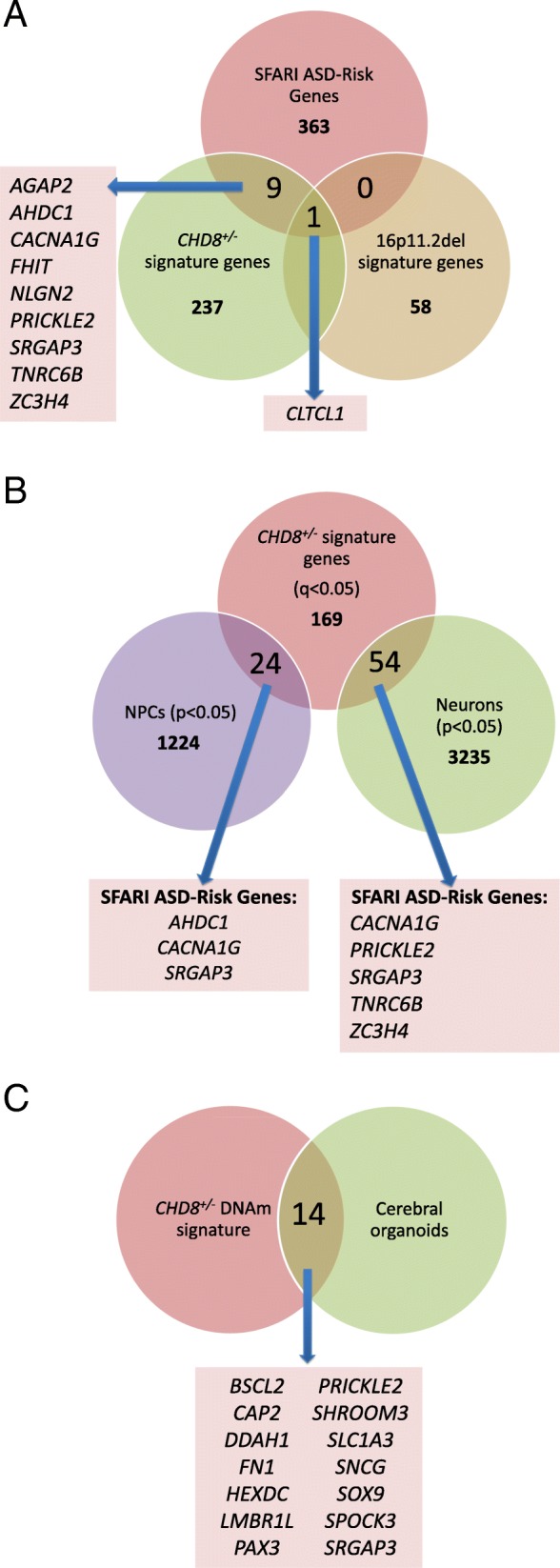
Results: DNAm patterns did not clearly distinguish heterogeneous ASD cases from controls. However, the homogeneous genetically-defined 16p11.2del and CHD8+/- subgroups each exhibited unique DNAm signatures that distinguished 16p11.2del or CHD8+/- individuals from each other and from heterogeneous ASD and control groups with high sensitivity and specificity. These signatures also classified additional 16p11.2del (n = 9) and CHD8 (n = 13) variants as pathogenic or benign. Our findings that DNAm alterations in each signature target unique genes in relevant biological pathways including neural development support their functional relevance. Furthermore, genes identified in our CHD8+/- DNAm signature in blood overlapped differentially expressed genes in CHD8+/- human-induced pluripotent cell-derived neurons and cerebral organoids from independent studies.
Conclusions: DNAm signatures can provide clinical utility complementary to next-generation sequencing in the interpretation of variants of unknown significance. Our study constitutes a novel approach for ASD risk-associated molecular classification that elucidates the vital cross-talk between genetics and epigenetics in the etiology of ASD.
Keywords: Autism spectrum disorder; DNA methylation; Epigenetics; Genetic stratification; Genomic variants; Heterogeneity.
Conflict of interest statement
The authors declare they have no competing interests.
Figures
References
-
- CDC . Centers for Disease Control and Prevention. 2014.
-
- Yuen RK, Thiruvahindrapuram B, Merico D, Walker S, Tammimies K, Hoang N, et al. Whole-genome sequencing of quartet families with autism spectrum disorder. Nature medicine. 2015;21(2):185–191. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
