

A real-time, click chemistry imaging approach reveals stimulus-specific subcellular locations of phospholipase D activity
- PMID: 31311871
- PMCID: PMC6681737
- DOI: 10.1073/pnas.1903949116
A real-time, click chemistry imaging approach reveals stimulus-specific subcellular locations of phospholipase D activity
Abstract
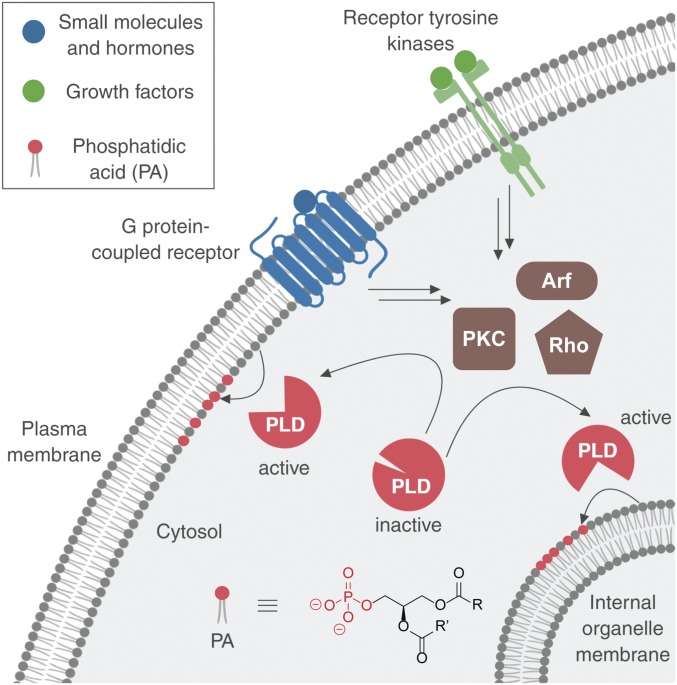
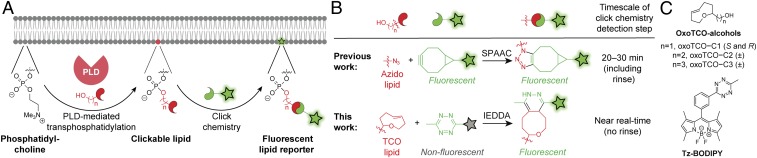
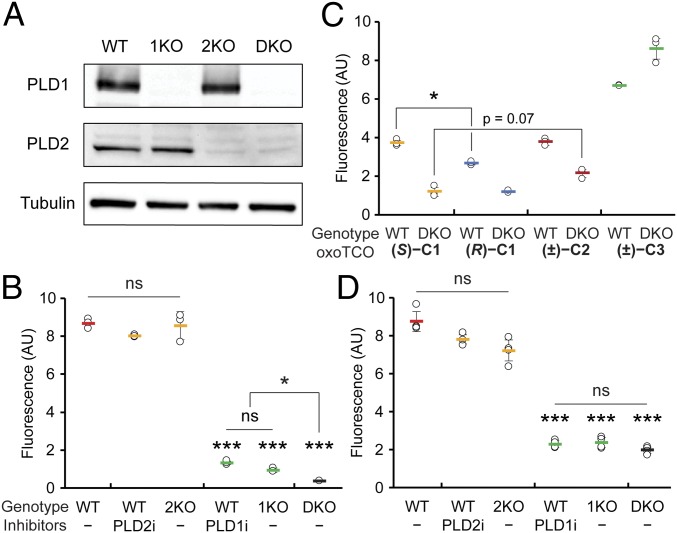
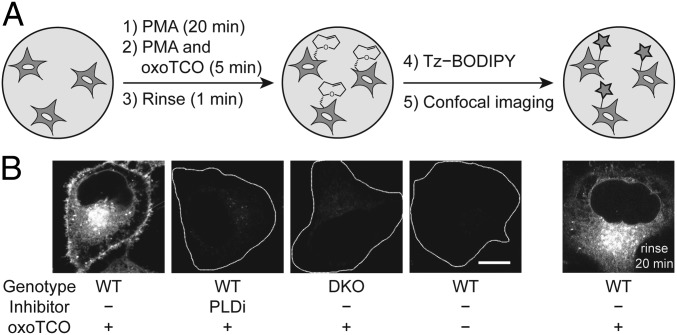
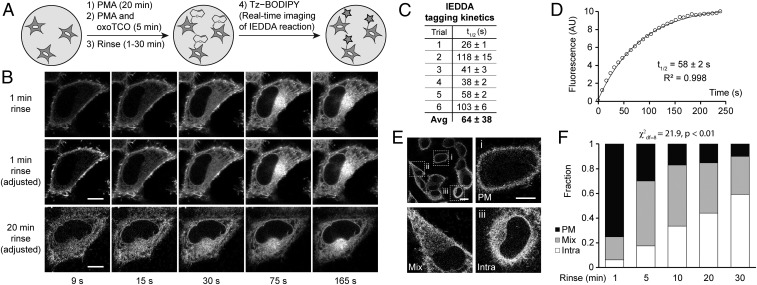
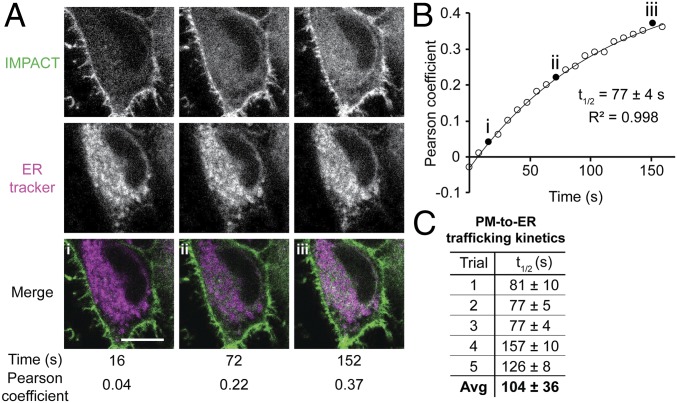
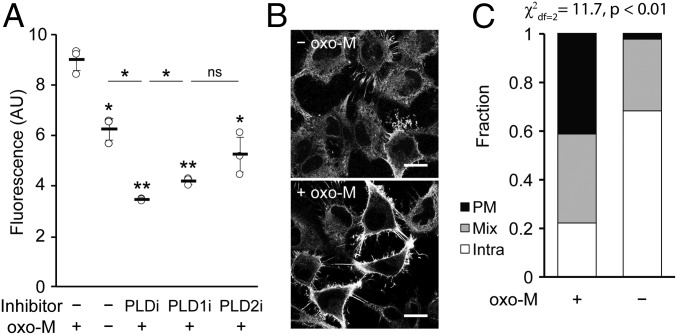
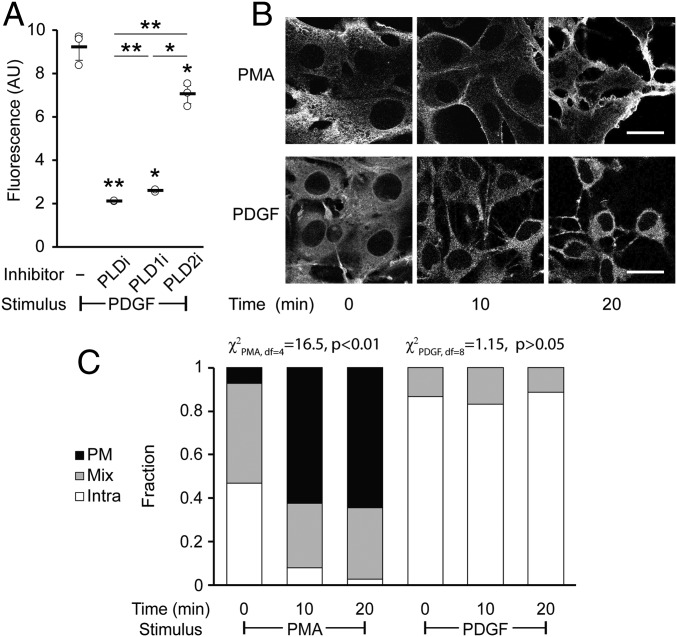
The fidelity of signal transduction requires spatiotemporal control of the production of signaling agents. Phosphatidic acid (PA) is a pleiotropic lipid second messenger whose modes of action differ based on upstream stimulus, biosynthetic source, and site of production. How cells regulate the local production of PA to effect diverse signaling outcomes remains elusive. Unlike other second messengers, sites of PA biosynthesis cannot be accurately visualized with subcellular precision. Here, we describe a rapid, chemoenzymatic approach for imaging physiological PA production by phospholipase D (PLD) enzymes. Our method capitalizes on the remarkable discovery that bulky, hydrophilic trans-cyclooctene-containing primary alcohols can supplant water as the nucleophile in the PLD active site in a transphosphatidylation reaction of PLD's lipid substrate, phosphatidylcholine. The resultant trans-cyclooctene-containing lipids are tagged with a fluorogenic tetrazine reagent via a no-rinse, inverse electron-demand Diels-Alder (IEDDA) reaction, enabling their immediate visualization by confocal microscopy in real time. Strikingly, the fluorescent reporter lipids initially produced at the plasma membrane (PM) induced by phorbol ester stimulation of PLD were rapidly internalized via apparent nonvesicular pathways rather than endocytosis, suggesting applications of this activity-based imaging toolset for probing mechanisms of intracellular phospholipid transport. By instead focusing on the initial 10 s of the IEDDA reaction, we precisely pinpointed the subcellular locations of endogenous PLD activity as elicited by physiological agonists of G protein-coupled receptor and receptor tyrosine kinase signaling. These tools hold promise to shed light on both lipid trafficking pathways and physiological and pathological effects of localized PLD signaling.
Keywords: click chemistry; lipid trafficking; phosphatidic acid; phospholipase D; second messengers.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Giepmans B. N. G., Adams S. R., Ellisman M. H., Tsien R. Y., The fluorescent toolbox for assessing protein location and function. Science 312, 217–224 (2006). - PubMed
-
- Liu Y., Su Y., Wang X., “Phosphatidic acid-mediated signaling” in Lipid-Mediated Protein Signaling (Springer Netherlands, Dordrecht, 2013), pp. 159–176. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
