

High-throughput targeted long-read single cell sequencing reveals the clonal and transcriptional landscape of lymphocytes
- PMID: 31311926
- PMCID: PMC6635368
- DOI: 10.1038/s41467-019-11049-4
High-throughput targeted long-read single cell sequencing reveals the clonal and transcriptional landscape of lymphocytes
Abstract
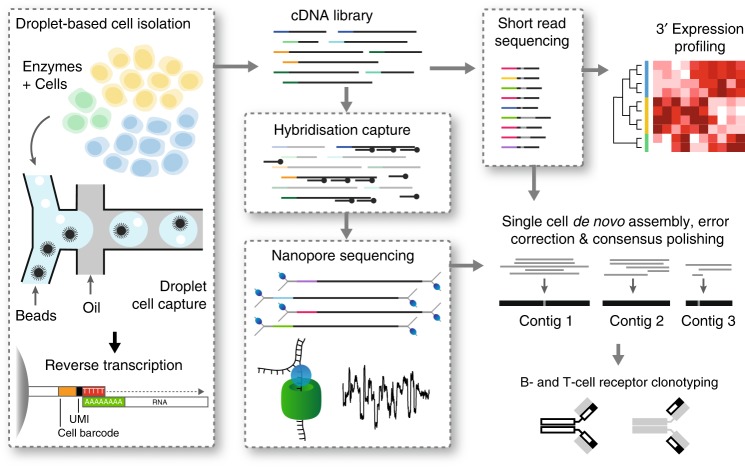
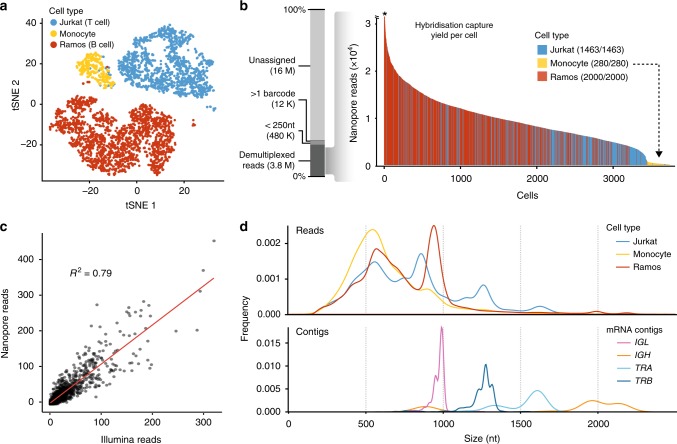
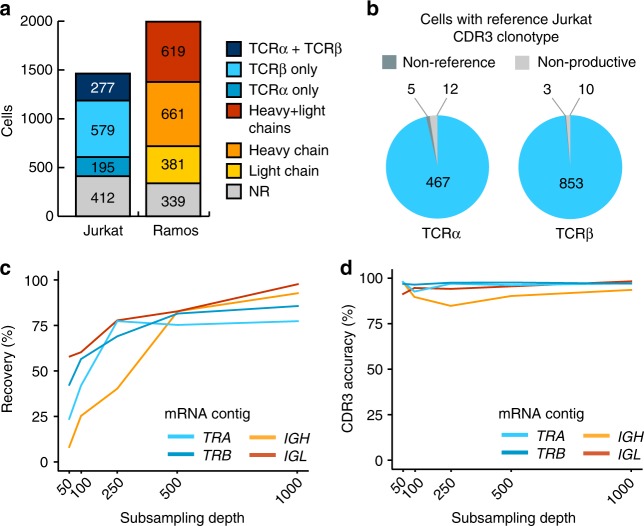
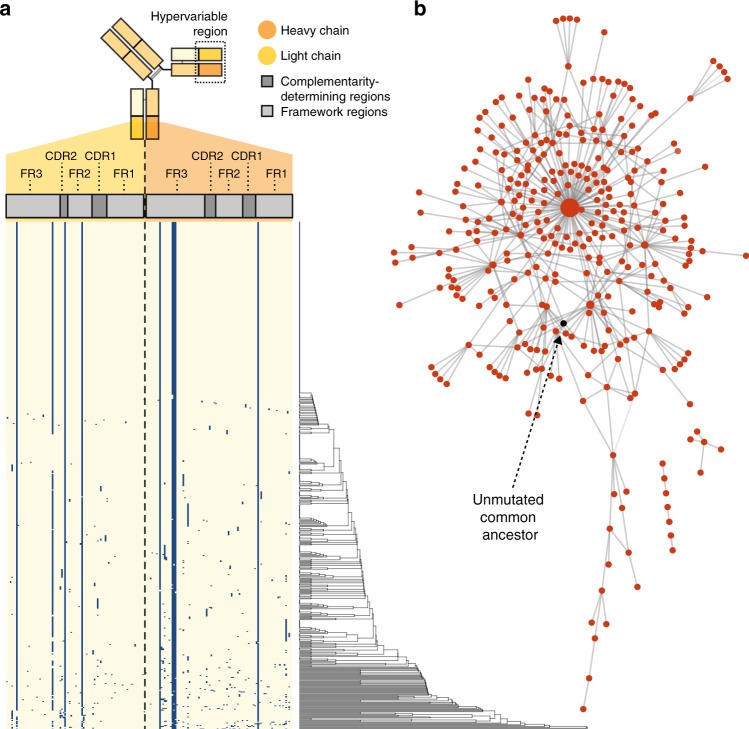
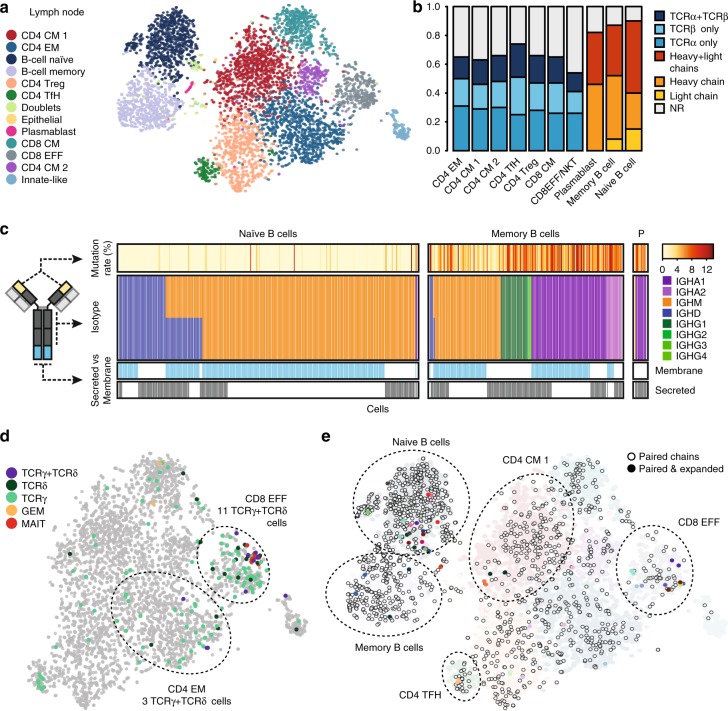
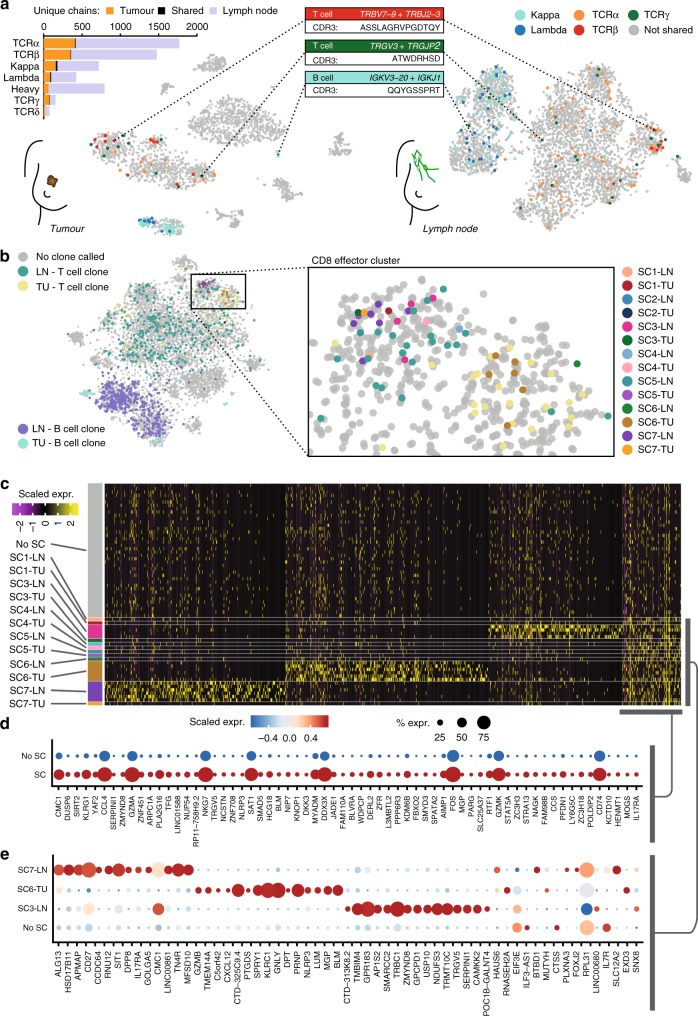
High-throughput single-cell RNA sequencing is a powerful technique but only generates short reads from one end of a cDNA template, limiting the reconstruction of highly diverse sequences such as antigen receptors. To overcome this limitation, we combined targeted capture and long-read sequencing of T-cell-receptor (TCR) and B-cell-receptor (BCR) mRNA transcripts with short-read transcriptome profiling of barcoded single-cell libraries generated by droplet-based partitioning. We show that Repertoire and Gene Expression by Sequencing (RAGE-Seq) can generate accurate full-length antigen receptor sequences at nucleotide resolution, infer B-cell clonal evolution and identify alternatively spliced BCR transcripts. We apply RAGE-Seq to 7138 cells sampled from the primary tumor and draining lymph node of a breast cancer patient to track transcriptome profiles of expanded lymphocyte clones across tissues. Our results demonstrate that RAGE-Seq is a powerful method for tracking the clonal evolution from large numbers of lymphocytes applicable to the study of immunity, autoimmunity and cancer.
Conflict of interest statement
M.S., G.A.A., C.C.G., S.L.C., J.M.F., K.J.L.J., M.A.S., and A.S. have filed a patent application covering some aspects of this work. The remaining authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
