

Defective HNF4alpha-dependent gene expression as a driver of hepatocellular failure in alcoholic hepatitis
- PMID: 31311938
- PMCID: PMC6635373
- DOI: 10.1038/s41467-019-11004-3
Defective HNF4alpha-dependent gene expression as a driver of hepatocellular failure in alcoholic hepatitis
Erratum in
-
Author Correction: Defective HNF4alpha-dependent gene expression as a driver of hepatocellular failure in alcoholic hepatitis.Nat Commun. 2023 Feb 10;14(1):757. doi: 10.1038/s41467-023-36548-3. Nat Commun. 2023. PMID: 36765087 Free PMC article. No abstract available.
Abstract
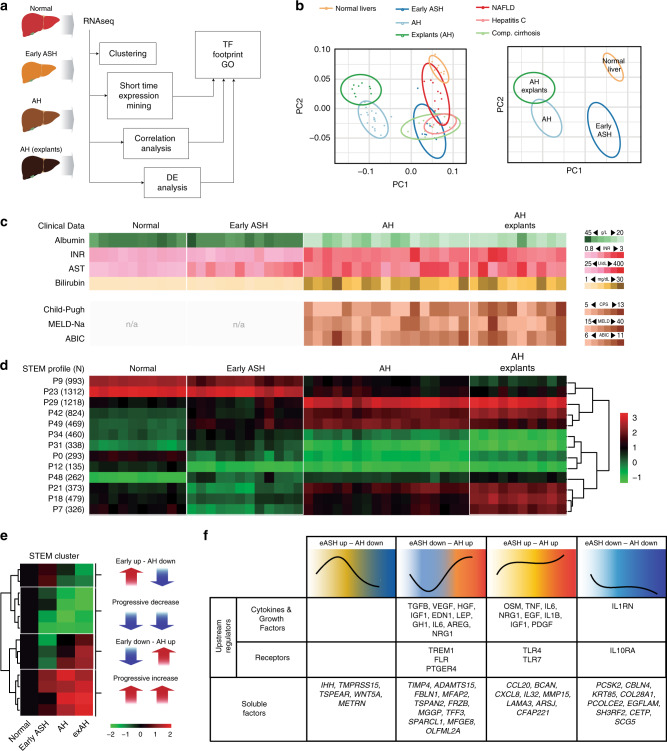
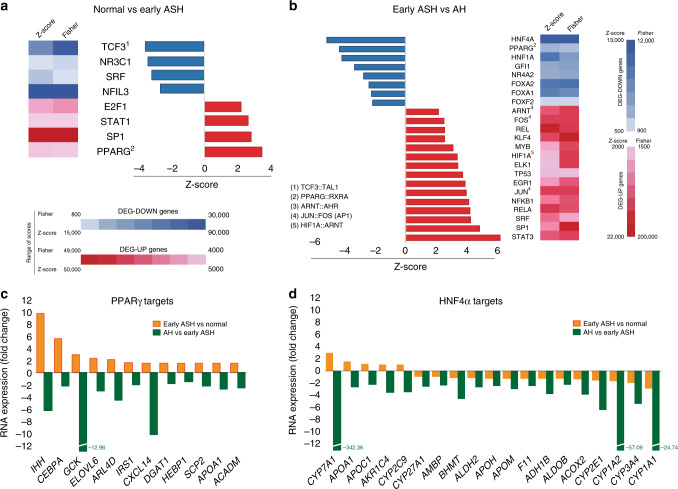
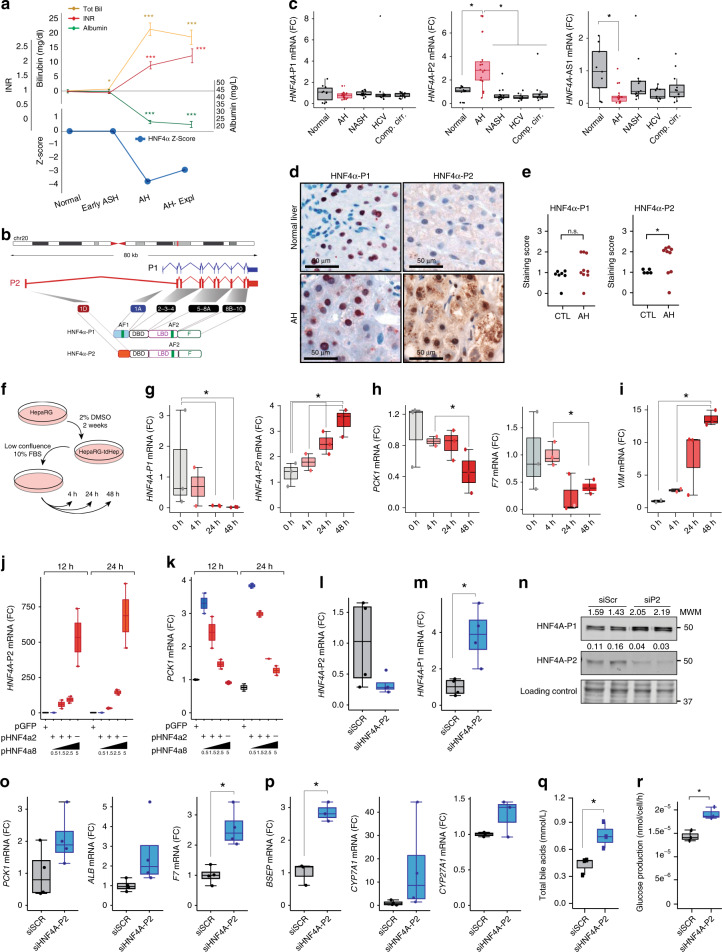
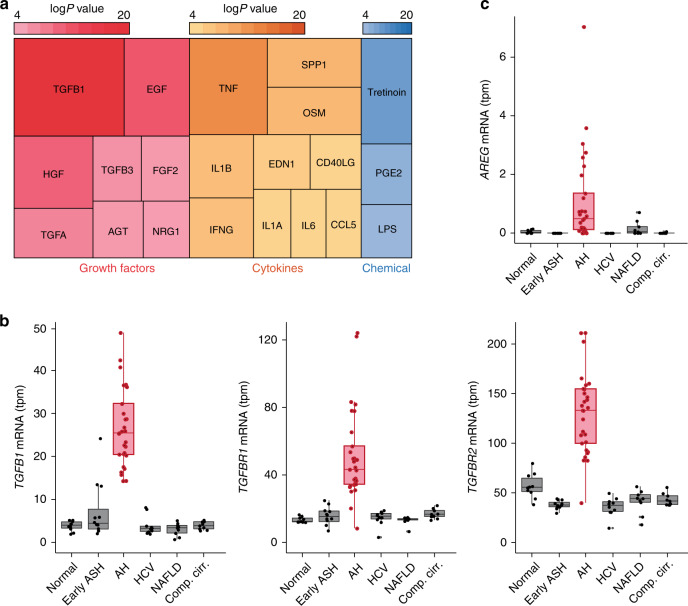
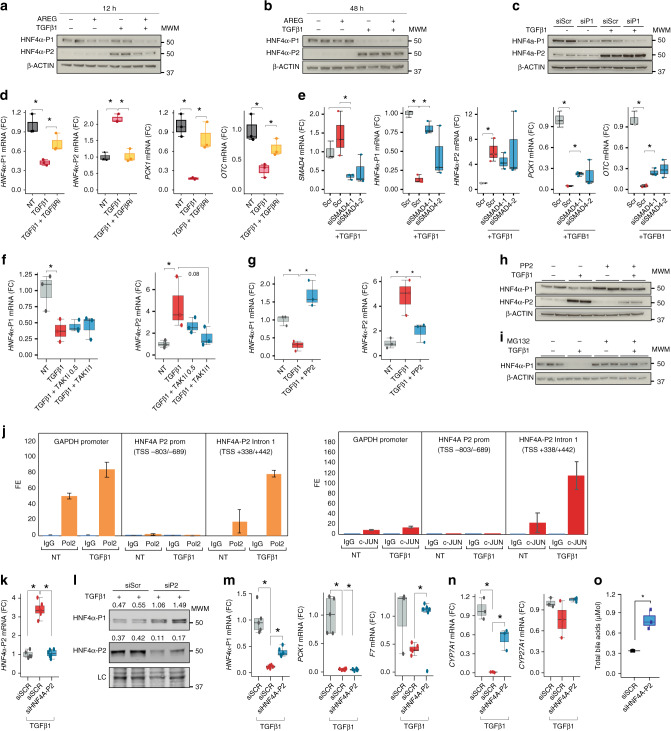
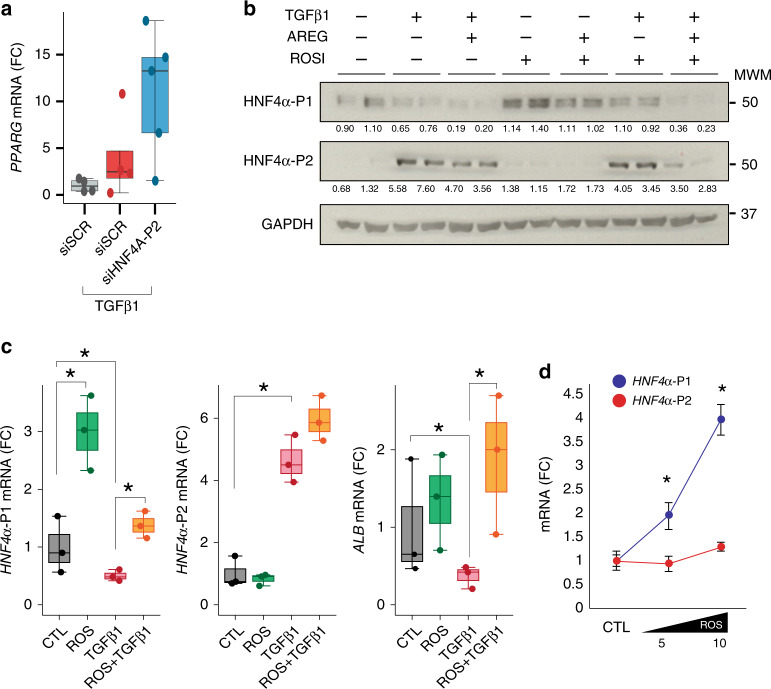
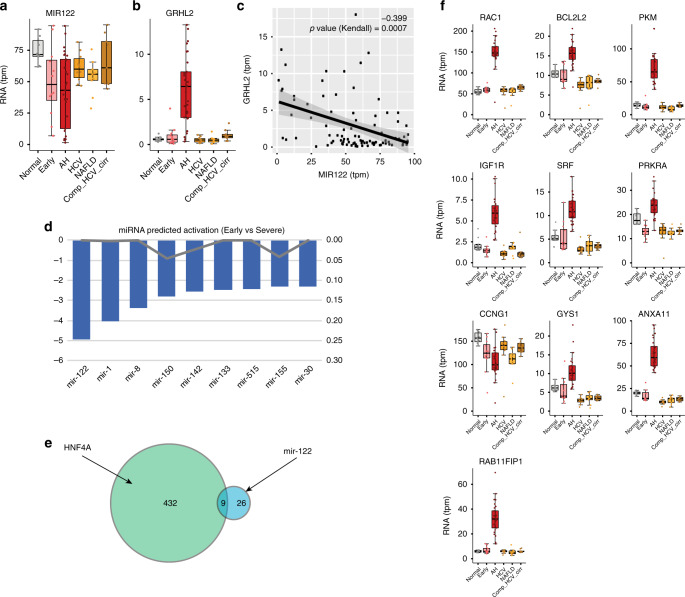
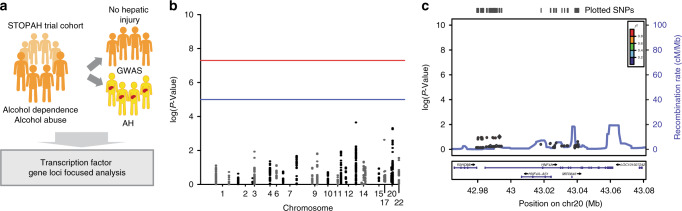
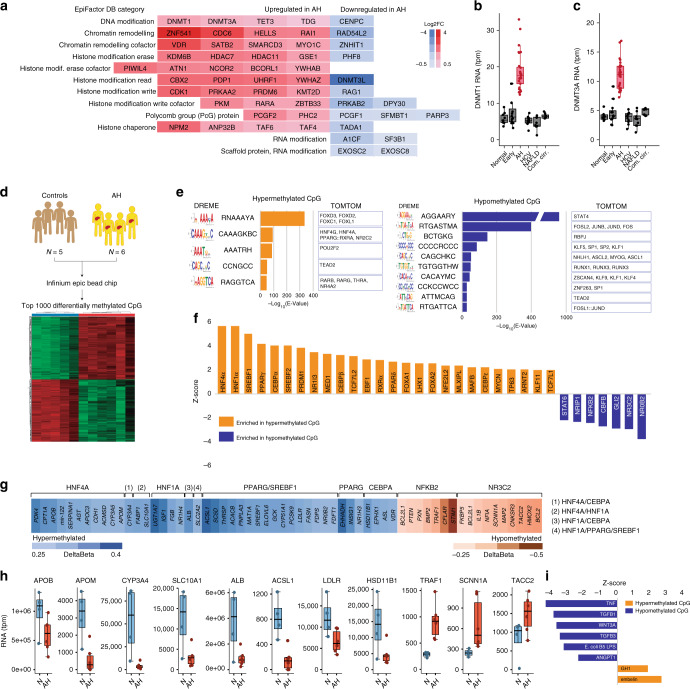
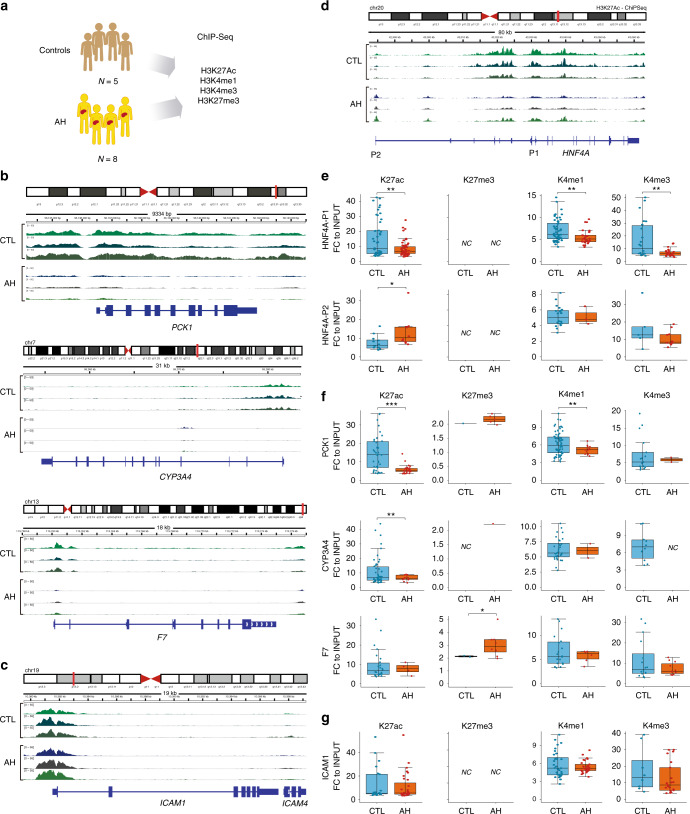
Alcoholic hepatitis (AH) is a life-threatening condition characterized by profound hepatocellular dysfunction for which targeted treatments are urgently needed. Identification of molecular drivers is hampered by the lack of suitable animal models. By performing RNA sequencing in livers from patients with different phenotypes of alcohol-related liver disease (ALD), we show that development of AH is characterized by defective activity of liver-enriched transcription factors (LETFs). TGFβ1 is a key upstream transcriptome regulator in AH and induces the use of HNF4α P2 promoter in hepatocytes, which results in defective metabolic and synthetic functions. Gene polymorphisms in LETFs including HNF4α are not associated with the development of AH. In contrast, epigenetic studies show that AH livers have profound changes in DNA methylation state and chromatin remodeling, affecting HNF4α-dependent gene expression. We conclude that targeting TGFβ1 and epigenetic drivers that modulate HNF4α-dependent gene expression could be beneficial to improve hepatocellular function in patients with AH.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Williams R, et al. Addressing liver disease in the UK: a blueprint for attaining excellence in health care and reducing premature mortality from lifestyle issues of excess consumption of alcohol, obesity, and viral hepatitis. Lancet. 2014;384:1953–1997. doi: 10.1016/S0140-6736(14)61838-9. - DOI - PubMed
-
- Mellinger Jessica L., Shedden Kerby, Winder Gerald Scott, Tapper Elliot, Adams Megan, Fontana Robert J, Volk Michael L., Blow Frederic C., Lok Anna S.F. The high burden of alcoholic cirrhosis in privately insured persons in the United States. Hepatology. 2018;68(3):872–882. doi: 10.1002/hep.29887. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 DK101426/DK/NIDDK NIH HHS/United States
- R01 DK116993/DK/NIDDK NIH HHS/United States
- MR/M003132/1/MRC_/Medical Research Council/United Kingdom
- R01 AA021171/AA/NIAAA NIH HHS/United States
- MR/R023026/1/MRC_/Medical Research Council/United Kingdom
- U01 AA018663/AA/NIAAA NIH HHS/United States
- R01 DK062277/DK/NIDDK NIH HHS/United States
- MR/R014019/1/MRC_/Medical Research Council/United Kingdom
- R01 DK109365/DK/NIDDK NIH HHS/United States
- R37 AA021171/AA/NIAAA NIH HHS/United States
- MR/K001949/1/MRC_/Medical Research Council/United Kingdom
- T32 DK007052/DK/NIDDK NIH HHS/United States
- R01 DK114012/DK/NIDDK NIH HHS/United States
- R01 AA023781/AA/NIAAA NIH HHS/United States
- U01 AA021908/AA/NIAAA NIH HHS/United States
- P30 DK120531/DK/NIDDK NIH HHS/United States
- UH3 TR000503/TR/NCATS NIH HHS/United States
- U01 AA026972/AA/NIAAA NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
