

Comparative performances of machine learning methods for classifying Crohn Disease patients using genome-wide genotyping data
- PMID: 31316157
- PMCID: PMC6637191
- DOI: 10.1038/s41598-019-46649-z
Comparative performances of machine learning methods for classifying Crohn Disease patients using genome-wide genotyping data
Abstract
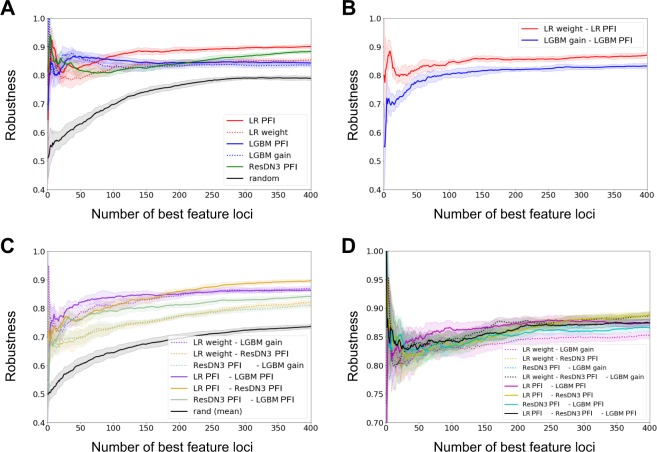
Crohn Disease (CD) is a complex genetic disorder for which more than 140 genes have been identified using genome wide association studies (GWAS). However, the genetic architecture of the trait remains largely unknown. The recent development of machine learning (ML) approaches incited us to apply them to classify healthy and diseased people according to their genomic information. The Immunochip dataset containing 18,227 CD patients and 34,050 healthy controls enrolled and genotyped by the international Inflammatory Bowel Disease genetic consortium (IIBDGC) has been re-analyzed using a set of ML methods: penalized logistic regression (LR), gradient boosted trees (GBT) and artificial neural networks (NN). The main score used to compare the methods was the Area Under the ROC Curve (AUC) statistics. The impact of quality control (QC), imputing and coding methods on LR results showed that QC methods and imputation of missing genotypes may artificially increase the scores. At the opposite, neither the patient/control ratio nor marker preselection or coding strategies significantly affected the results. LR methods, including Lasso, Ridge and ElasticNet provided similar results with a maximum AUC of 0.80. GBT methods like XGBoost, LightGBM and CatBoost, together with dense NN with one or more hidden layers, provided similar AUC values, suggesting limited epistatic effects in the genetic architecture of the trait. ML methods detected near all the genetic variants previously identified by GWAS among the best predictors plus additional predictors with lower effects. The robustness and complementarity of the different methods are also studied. Compared to LR, non-linear models such as GBT or NN may provide robust complementary approaches to identify and classify genetic markers.
Conflict of interest statement
The authors declare no competing interests.
Figures
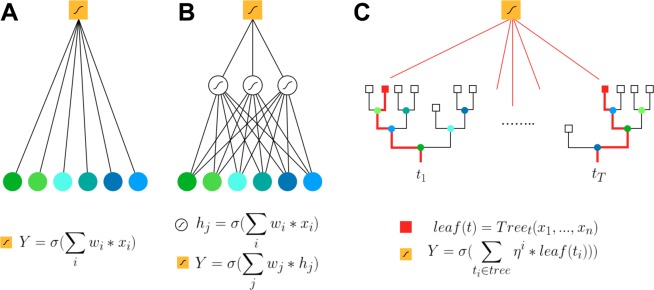
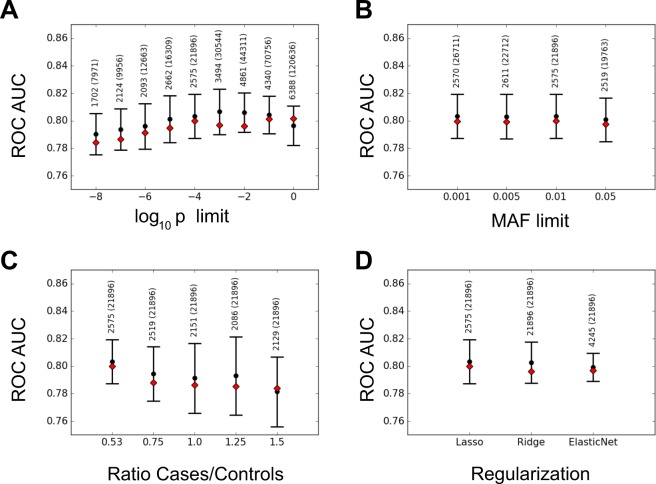
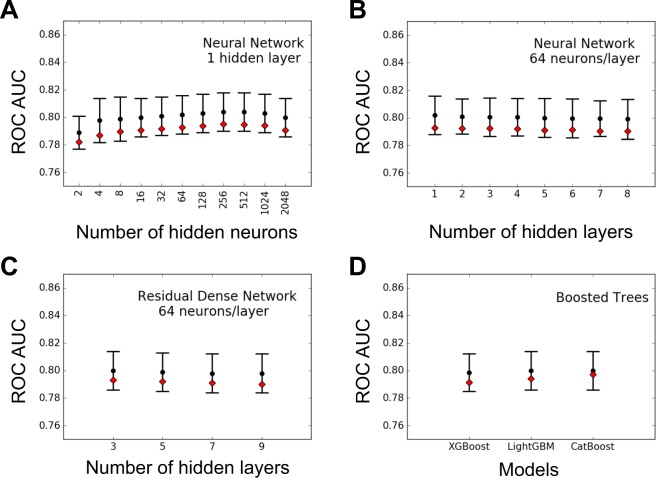
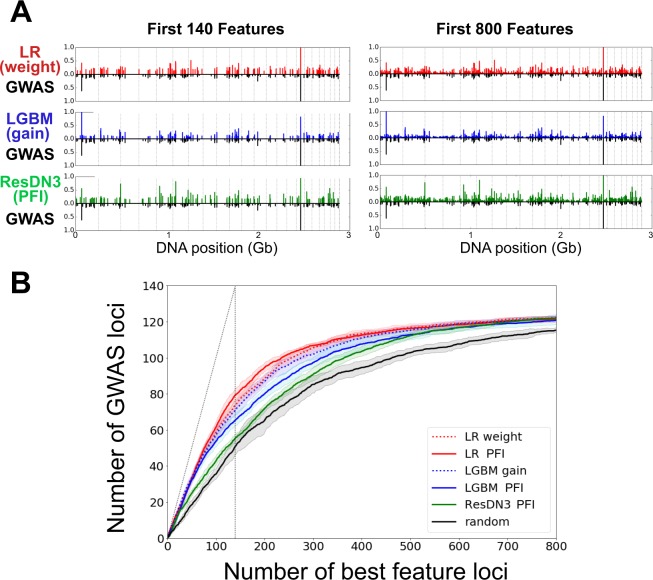
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
